Mitte August habe ich den reichweitestärksten Tweet meiner bisherigen Social-Media-„Karriere“ und zugleich den erfolgreichsten deutschsprachigen Wissenschaftskommunikations-Tweet der Woche geschrieben. Verrückterweise wurde er wohl auch wegen des süßen Babyfotos so oft geliket und retweetet, dessen Verwendung ich gerade kritisierte:
Längst nicht jeder wird den ernsten Hintergrund dieses Tweets verstanden haben oder nachvollziehen können – aber es waren auch viele WissenschaftlerInnen und WissenschaftskommunikatorInnen dabei, und einige Rückmeldungen haben mir gezeigt, dass ich mit meinem Frust nicht alleine bin: Seit Jahren werden uns sowohl in der Fachpresse als auch in der Laienpresse Ergebnisse von Mikrobiom-Sudien an Tiermodellen so verkauft, als gälten sie eins zu eins auch für Menschen. Mal sind es die Studienpublikationen selbst, die das in der Überschrift suggerieren und erst irgendwo in der Einleitung oder gar im Methodenteil klarstellen, dass man an einem bestimmten Mäusestamm gearbeitet hat. Mal sind es die Pressemitteilungen der Forschungseinrichtung, die diesen Umstand erst gegen Ende beiläufig erwähnen und zugleich durch das mitgelieferte Bildmaterial falsche Erwartungen wecken, wie in diesem Fall. Und mal fallen die Mäuse und Ratten erst beim Transfer der Nachricht in die Tagespresse unter den Tisch.
Es sind nicht nur Provinzblätter und Werbeseiten, die falsche Erwartungen wecken: Ich habe mich hier schon einmal über eine krasse Text-Bild-Schere im News-Teil des renommierten Wissenschaftsjournals Science mokiert, in dem eine Studie zu Unterschieden zwischen der Darmflora männlicher und weiblicher Mäuse und damit einhergehenden Neigungen zu Autoimmunerkrankungen mit einer Illustration aufgehübscht wurde, in der eine Frau und ein Mann zu sehen sind.
Bei Twitter hat mich dann prompt jemand belehrt: Mäuse und Menschen seien als Säugetiere so eng verwandt und einander physiologisch so ähnlich, dass man an Mäusen gewonnene Erkenntnisse über irgendwelche Mechanismen und Signalwege an der Darmwand durchaus auf Menschen übertragen könne. I beg to differ, und das möchte ich hier anhand zweier aktueller Übersichtsarbeiten näher ausführen – in Ergänzung dessen, was ich bereits vor drei Jahren über Mäuse schrieb (Live fast, Love hard, Die young):
Nguyen, T. L. A., Vieira-Silva, S., Liston, A., & Raes, J. (2015). How informative is the mouse for human gut microbiota research? Disease Models & Mechanisms, 8(1), 1–16. http://doi.org/10.1242/dmm.017400
Hugenholtz, F., & de Vos, W. M. (2018). Mouse models for human intestinal microbiota research: a critical evaluation. Cellular and Molecular Life Sciences, 75(1), 149–160. http://doi.org/10.1007/s00018-017-2693-8
Ja, anatomisch und physiologisch haben Mäuse und wir vieles gemeinsam. Aber es gibt auch biologische Unterschiede: im Genom, in der Ernährung, in der Anatomie und Physiologie des Verdauungstrakts und seiner Teile (einschließlich des örtlichen Immunsystems), in der Zusammensetzung der Magen- und Darmflora und in den krankhaften Veränderungen dieses Mikrobioms.
Genom
Mit Mäusen meine ich im Folgenden Stämme der Art Mus musculus, die zum Teil seit über 100 Jahren als Versuchstiere gezüchtet werden. (Als Haustiere werden sogenannte Farbmäuse schon seit 1200 v. Chr. kultiviert, anfangs in China.) Es gibt über 400 Zuchtstämme.
Der letzte gemeinsame Vorfahr von Maus und Mensch lebte vor über 90 Millionen Jahren. Dennoch stimmen wegen einer starken Konservierung (also Selektionsvorteilen der alten Sequenzen gegenüber neuen Varianten, die durch Mutation entstehen) über 85 Prozent ihres Genoms noch überein. Die größten Unterschiede finden sich nicht in DNA-Abschnitten, die Proteine codieren, sondern in Steuerungssequenzen wie den Bindungsstellen von Transkriptionsfaktoren.
Insbesondere das Immunsystem und seine Regulierung haben sich zwischen Maus und Mensch stark auseinander entwickelt. Die Unterschiede im lokalen Immunsystem des Verdauungstrakts dürften einer der Gründe dafür sein, dass die Ergebnisse vieler an Mäusen durchgeführten Studien zu Entzündungen und Erkrankungen mit Beteiligung des Immunsystems bei Menschen nicht reproduziert werden konnten.
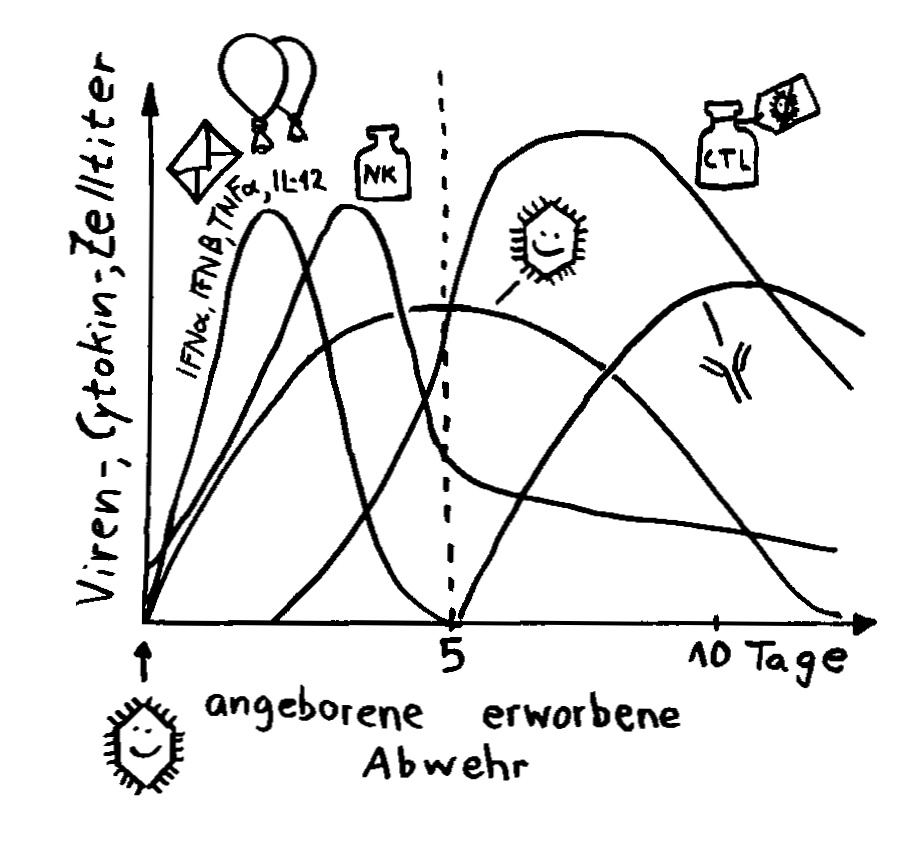
Das Gen für TLR5, jenen Rezeptor der angeborenen Abwehr, um den es in der Nature-Veröffentlichung von Fulde et al. geht, die mit dem süßen Baby „beworben“ wurde, gibt es sowohl bei Menschen als auch bei Mäusen. Überhaupt ähneln sich die TLR-Repertoires beider Arten – identisch aber sind sie nicht. Es ist auch nicht sicher, dass die einander genetisch entsprechenden Rezeptoren in Maus und Mensch exakt dieselben Funktionen ausüben, in denselben Zelltypen zu denselben Zeiten exprimiert werden, dieselben Signalketten auslösen und so weiter.
Ernährung, Energie- und Vitamin-Gewinnung
Mäuse sind Allesfresser, wobei der Großteil ihrer Kost pflanzlich ist. Ihre Nahrung enthält wesentlich mehr schwer aufzuschließende Kohlenhydrate als unsere. Menschen sind im Prinzip ebenfalls Omnivoren, die aber weniger schwer verdauliche Pflanzenbestandteile zu sich nehmen. Auch der Aufbau des Verdauungstrakts strikter Veganer ist evolutionär an die gemischte, fleischhaltige Kost ihrer Urahnen angepasst. Unsere Darmflora reagiert dagegen zügig (wenn auch mit recht subtilen Anpassungen) auf eine Ernährungsumstellung.
Ein Problem bei Mäuse-Studien: Die Zusammensetzung des Trockenfutters wird von den Herstellern nicht offengelegt und schwankt offenbar je nach Agrarmarktlage. Manchmal enthält es beispielsweise Luzerne, die wiederum Phytoestrogene enthalten kann. Diese Substanzen können im Körper wie Estrogen wirken und damit etwa Immunreaktionen oder auch die Zusammensetzung des Mikrobioms beeinflussen.
Die Transitzeit einer Mahlzeit beträgt beim Menschen 14-76 Stunden – je schwerer verdaulich, desto länger. Resistente Stärke ist zum Beispiel fast 20 Stunden länger in uns unterwegs als leicht verdauliche Stärke. Bei Mäusen ist die Transitzeit mit 6-7 Stunden deutlich kürzer: Wie alle kleinen Warmblüter haben sie eine viel höhere Stoffwechselrate und daher einen (relativ zum Körpergewicht) viel größeren Stoffumsatz als wir. Sie müssen fast rund um die Uhr fressen, um ihren Energiebedarf zu stillen – und haben daher nur wenige Stunden Zeit, ihre schwer verdauliche Nahrung aufzuschließen. Sie lösen dieses Problem mit einem Trick, den wir Menschen (zum Glück!) nicht beherrschen.
Im vorderen Bereich des Mäuse-Dickdarms gibt es eine „Schleimfalle“: Falten und Furchen, in denen mit Darmbakterien durchmischter Nahrungsbrei hängen bleibt. Von dort wird er ein Stück „stromaufwärts“ in den Blinddarm geschoben. In dieser Fermentierkammer gewinnen die Bakterien Fettsäuren, Vitamin K und einige B-Vitamine aus der Kost. Die Nährstoffe und Vitamine können im Dickdarm nicht resorbiert werden und werden mit dem Kot ausgeschieden. Aber Mäuse fressen ihren Kot (sogenannte Koprophagie) und nehmen die wertvollen Stoffe beim zweiten Durchlauf im Dünndarm ins Blut auf. Auch ein Teil der wertvollen Darmflora wird so recycelt.
Aufbau des Verdauungstrakts
Mäuse haben – anders als wir – einen großen drüsenfreien Vormagen, der als reiner Nahrungsspeicher dient und mit einem pH-Wert von 3 bis 4 weniger sauer ist als der menschliche Magen mit seinem pH-Wert von etwa 1. In diesem weniger aggressiven Milieu gedeihen Bakterien: Die Wand des Vormagens ist mit einem Biofilm aus Lactobacillus-Arten ausgekleidet. Auch der Drüsenmagen, der sich an diese Kammer anschließt, ist weniger sauer als der menschliche Magen, da sich in ihm ständig frische Kost mit den Magensäften und der älteren Kost vermischt. Im menschlichen Magen überleben nur wenige Bakterien, die sich an die starke Säure angepasst haben: Streptokokken, Prevotella und Helicobacter pylori.
Der Dünndarm ist bei beiden Arten der längste Teil des Verdauungstrakts. Mit 33 cm ist er bei der Maus 2,5-mal so lang wie der Dickdarm, beim Menschen mit 700 cm 7-mal so lang. Noch deutlicher werden die Verhältnisse bei der Betrachtung der Flächen: In der Maus hat der Dünndarm eine 18-mal größere Oberfläche als der Dickdarm, beim Menschen beträgt der Faktor sogar 400. Durch diese riesige Grenzschicht wird ein Großteil der Nährstoffe in den Körper aufgenommen.
Die Schleimhaut des Dünndarms ist bei der Maus glatt, beim Menschen wirft sie ringförmige Falten, die die Oberfläche vergrößern und den im Schleim lebenden Bakterien Nischen bieten. Die Zotten oder Villi, die ebenfalls die Oberfläche vergrößern, sind dafür bei der Maus länger als beim Menschen.
Der Dickdarm einer Maus ist bis zu 14 cm lang, der eines Menschen etwa 105 cm – relativ zur Körpermasse also viel kürzer als bei dem kleinen, leichten Nager. Man unterscheidet Blinddarm (Caecum – nicht zu verwechseln mit dem Wurmfortsatz, der von Laien oft als Blinddarm bezeichnet wird) und Grimmdarm (Colon). Der Mäuse-Blinddarm dient, wie im vorigen Abschnitt erwähnt, als Fermentationskammer und ist mit 3 bis 4 cm ziemlich lang. Beim Menschen findet die Fermentation dagegen nur im Colon statt; der etwa 6 cm kurze Blinddarm hat keine wichtige Funktion. Der Wurmfortsatz ist bei Mäusen nicht so klar vom Blinddarm abgegrenzt wie wie bei uns. Der Grimmdarm ist bei der Maus glattwandig, beim Menschen hat er Ausbuchtungen (Hausten genannt).
Die Becherzellen, die den Darmschleim produzieren, konzentrieren sich bei der Maus auf den Dünndarm und den Anfang des Dickdarms, während sie sich beim Menschen bis hinunter zum Rektum über die ganze Länge verteilen. Die Paneth-Zellen, die antibakterielle Produkte wie die Defensine ausschütten, fehlen bei der Maus im Colon; es gibt sie nur im Blinddarm. Beim Menschen finden sich dagegen auch einige im Anfang des Colons. Auch die Menge, die Speicherung und die Ausschüttung von Defensinen unterscheiden sich zwischen den Arten; das wiederum kann über die Regulierung der örtlichen Immunreaktionen die Zusammensetzung des Mikrobioms beeinflussen.
Die Colon-Schleimhaut des Menschen produziert den Schleim schneller (etwa 240 µm/h) als die der Maus (etwa 100 µm/h). Die Colon-Schleimschicht wird beim Menschen etwa 480 µm dick und bei der Maus etwa 190 µm. Der Schleim hat eine ähnliche Zähigkeit und Porosität und besteht aus ähnlichen Schleimproteinen, die allerdings in beiden Arten anders glykosyliert werden. Die unterschiedlichen Glykane, die dabei seitlich an das Protein-Grundgerüst angehängt werden, sodass das Makromolekül schließlich wie eine Flaschenbürste aussieht, beeinflussen wiederum die Selektion der Darmflora.
Zusammensetzung des Mikrobioms
Die Darmflora von Maus und Mensch wird von zwei Bakterienstämmen (Stämmen im Sinne von phyla, nicht strains) dominiert, den Bacteroidetes und den Firmicutes. Das gilt auch für viele andere Säugetiere, egal ob Pflanzen- oder Fleischfresser. Dennoch gibt es beträchtliche Unterschiede.
Um diese Unterschiede zu entdecken, muss man sich auf Bakterien konzentrieren, die bei der Mehrheit der untersuchten Mäuse bzw. Menschen vorkommen, und die „Ferner-liefen-Bakterien“ ausklammern, die zwar zur Diversität des Mikrobioms einer der Art beitragen, aber nur in einem Bruchteil der untersuchten Individuen nachzuweisen sind. Sogenannte metagenomische Analysen haben gezeigt: Von den 60 Gattungen der Kern-Darmflora von Mäusen gehören 25 auch zum Kernbestand im menschlichen Darm. Allerdings haben nur 4 Prozent der Mäuse-Bakteriengene mehr oder weniger identische Entsprechungen im Pool der Menschen-Bakteriengene. Ein Beispiel: Lactobacillus reuteri kommt sowohl in Mäusen als auch in Menschen vor, aber die Stämme in den Mäusen (jetzt im Sinne von strains – verdammte terminologische Ambivalenz!) haben andere Urease-Gene, die diese Enzyme befähigen, in einem sauren Milieu zu leben. Auf der funktionellen Ebene sind die Unterschiede kleiner: 80 Prozent der in den Metagenomik-Datenbanken verzeichneten Gen- bzw. Protein-Funktionen sind sowohl bei der Maus als auch beim Menschen vertreten.
Auch wenn eine Bakterien-Gattung bei Mensch und Maus vertreten ist, kann sie in einer der Arten dominieren und in der anderen eine Randerscheinung bleiben. Im Mäuse-Dünndarm sind Faecalibacterium, Prevotella und Ruminococcus viel seltener als im menschlichen Dünndarm. Dafür sind Turicibacter, Alistipes und Lactobacillus bei Mäusen viel dominanter als bei uns. Die Gattungen Clostridium, Bacteroides und Blautia sind in beiden Arten etwa gleich stark vertreten.
Wie hier im Blog schon mehrfach besprochen, prägen die sogenannten segmentierten filamentösen Bakterien (SFB) im Darm von Mäusen die Reifung des Immunsystems – vor allem, indem sie in der Schleimhaut junger Mäuse die Bildung von entzündungsfördernden Th17-Helferzellen auslösen. Das prägt nicht nur die „Stimmung“ des örtlichen Immunsystems, sondern sogar die Entwicklung des Gehirns. Bis vor wenigen Jahren dachte man, im menschlichen Darm gebe es gar keine SFB. Inzwischen wurden diese Bakterien, die zu den Firmicutes zählen und auch als Candidatus arthromitus bezeichnet werden, im Mikrobiom einiger (aber längst nicht aller) Kleinkinder unter drei Jahren entdeckt. Ob sie dort eine ähnlich prägende Rolle spielen wie in jungen Mäusen und so womöglich die Neigung bestimmter Erwachsener zu chronischen Entzündungen fördern, ist noch unklar.
Erschwert werden Vergleiche zwischen Mensch und Maus durch die enormen Mikrobiom-Unterschiede zwischen den untersuchten Mäusen. Nicht nur der Zuchtstamm, sondern auch ihr Futter, die Einrichtung, in der sie gehalten werden, und der Käfig, in dem sie mit anderen Mäusen zusammenleben, prägen die Zusammensetzung. (Man denke an die Koprophagie!)
Enterotypen
Auch beim Menschen unterscheidet sich die Mikrobiom-Zusammensetzung zwischen den Individuen. Seit einigen Jahren kennt man drei Enterotypen: Gruppen, deren Darmflora von jeweils anderen Bakterien dominiert wird. Wie klar und stabil diese Gruppen voneinander abgegrenzt sind, ist allerdings umstritten, und wie sie zustande kommen, ist unbekannt.
Bei Labormäusen wurden bislang zwei Enterotypen identifiziert: Wenn Lachnospiraceae und Ruminococcaceae dominieren, entspricht dies dem menschlichen Enterotyp 3; wenn Bacteroidaceae und Enterobacteriaceae vorherrschen, ähnelt dies dem menschlichen Enterotyp 1. Auch bei Wildmäusen lassen sich zwei Enterotypen unterscheiden, die von Bacteroides oder Robinsoniella dominiert werden.
Bei den Labormäusen korreliert die Einteilung mit dem Artenreichtum des Mikrobioms und mit der Neigung zu Entzündungen. Der Bacteroidaceae/Enterobacteriaceae-Enterotyp ist artenärmer und weist mehr Calciprotectin auf, das als Entzündungsmarker fungiert. Das entspricht den Verhältnissen bei Menschen mit starkem Übergewicht, deren Darmflora ebenfalls verarmt und durch ähnliche Bakteriengruppen (Bacteroidetes und Proteobacteria) dominiert ist und die ebenfalls stärker zu Entzündungen neigen.
Krankhafte Veränderungen
Während sich das Mikrobiom in Maus-Modellen für Fettleibigkeit auf ähnliche Weise verschiebt wie beim Menschen, sind die Parallelen bei anderen Erkrankungen längst nicht so stark. So kann zum Beispiel nach wie vor kein Modell für Colitis ulcerosa alle wichtigen Eigenschaften des Erkrankungsprozesses und der Darmflora-Veränderung rekapitulieren.
Das führt auch zum Scheitern von Therapie-Ansätzen. So hatte man nach Studien an IL-10-Knockout-Mäusen große Hoffnungen, dass das Zytokin IL-10 chronisch-entzündliche Darmerkrankungen eindämmen könne. In klinischen Studien an Menschen ließ sich der Effekt aber nicht reproduzieren – vermutlich, weil Menschen einen großen Pool recht unterschiedlicher IL-10-Rezeptoren haben.
Mäuse mit humanisierter Darmflora: keine Patentlösung
Angesichts der Unterschiede zwischen den Darmfloren von Maus und Mensch und der Unvollkommenheit, mit der viele Tiermodelle menschliche Erkrankungen imitieren, liegt es nahe, das Mikrobiom der Mäuse menschenähnlicher zu machen. Dazu kann man keimfreie, also ohne eigenes Mikrobiom geborene und gehaltene junge Mäuse mit menschlicher Darmflora animpfen. Man spricht dann von humanisierten gnotobiotischen Mäusen – „gnotobiotisch“, weil man dann weiß, welche Bakterien in ihnen leben (griechisch gnosis = Wissen).
Dabei können sich alle der in der menschlichen Darmflora vorkommenden Stämme (Phyla), 11 von 12 der Klassen und etwa 88 Prozent der Gattungen aus dem humanen Mikrobiom im Mäusedarm ansiedeln: gar keine schlechte Annäherung. Aber dieses aus dem Menschen stammende Mikrobiom und die Maus haben keine gemeinsame Evolution durchlaufen, sie haben sich nicht über Jahrmillionen aneinander anpassen können. Und wie sich zeigt, reifen humanisierte gnotobiotische Mäuse nicht normal; sie reagieren zum Beispiel nicht normal auf Infektionen. Vielleicht liegt es daran, dass Bakterien und Mäusezellen nicht genau dieselbe Sprache sprechen, ihre Botenstoffe und Signalketten also wegen der 90 Millionen Jahre getrennter Evolution von Maus und Mensch nicht mehr zueinander passen. Oder bei der Ansiedlung gehen einige seltene, aber für die Entwicklung essentielle Bakterien verloren.
An Mäusen führt kein Weg vorbei
All das heißt nicht, dass man keine Mikrobiom-Forschung oder keine immunologischen Studien an Mäusen betreiben sollte. Mäuse sind klein, haben eine kurze Generationsdauer und sind günstig in der Anschaffung und im Unterhalt. Man kann sie auch genetisch verändern, um z. B. bestimmte Gene „auf Knopfdruck“ auszuschalten (sog. Knockout-Mäuse). Für viele Versuche müssen sie getötet werden, etwa um ihnen Gewebeproben zu entnehmen – und zwar in großer Zahl, um statistisch belastbare Ergebnisse zu erhalten. Dieselben Untersuchungen etwa an Schweinen oder Affen durchzuführen, wäre ethisch und praktisch problematisch. Grundlegende Mechanismen oder Signalwege lassen sich an Mäusen durchaus ermitteln – aber sie müssen gründlich am Menschen überprüft werden.
Forscherinnen und Wissenschaftskommunikatoren sollten der Versuchung eigener vorschneller Extrapolationen und erst recht mutwillig evozierter Missverständnisse widerstehen: Wer an Mäusen geforscht hat, sollte das bereits in der Überschrift und im Abstract deutlich machen. Und die Menschen in den PR-Abteilungen der Forschungseinrichtungen sollten wirklich die Finger von süßen Babyfotos und Formulierungen wie „Kindheit“ lassen, wenn es um junge Mäuse geht. Auch der inflationäre Gebrauch von Superlativen, mit denen die jeweilige Studie aus dem medialen Grundrauschen herausgehoben werden soll, geht letzten Endes nach hinten los: Wenn ich innerhalb einer Woche lese, dank der bahnbrechenden Studie A sei nun endlich bewiesen, dass die Darmflora in einem kleinen Zeitfenster nach der Geburt fürs ganze Leben geprägt werde, und die bahnbrechende Studie B habe endlich gezeigt, dass anhaltender Durchfall bei Erwachsenen die Darmflora nachhaltig verändern könne, dann werde ich nächste Woche die bahnbrechenden Studien C, D und E mit einem Achselzucken an mir vorüberziehen lassen.