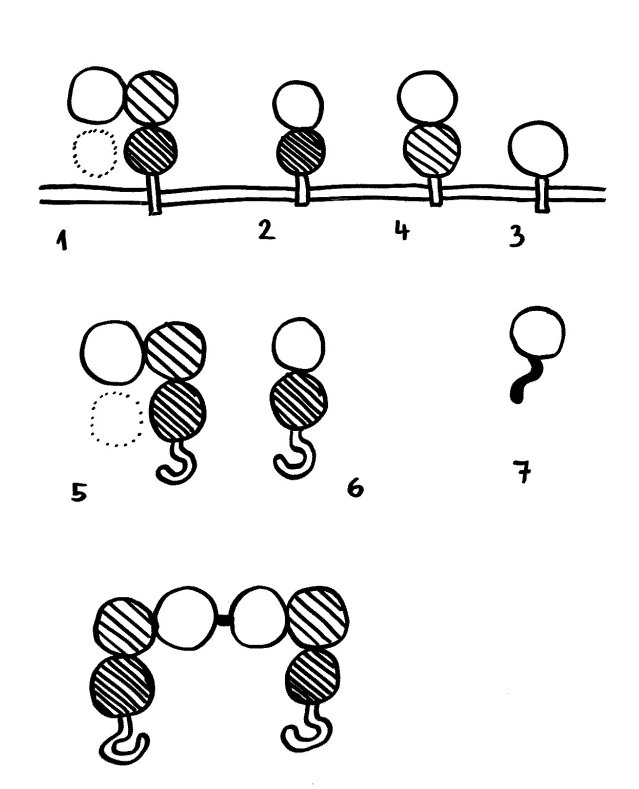
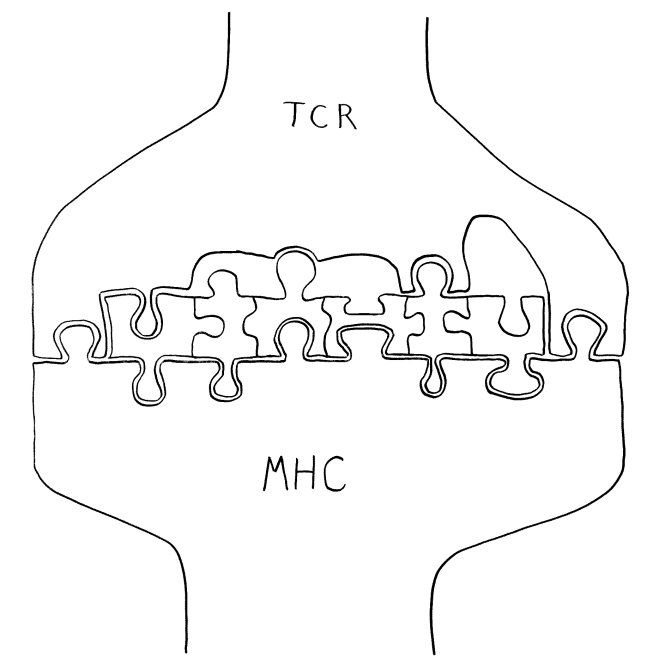
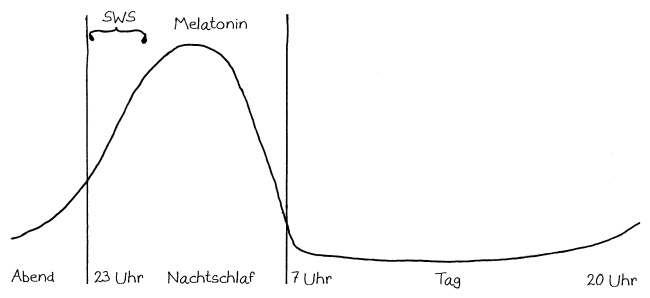
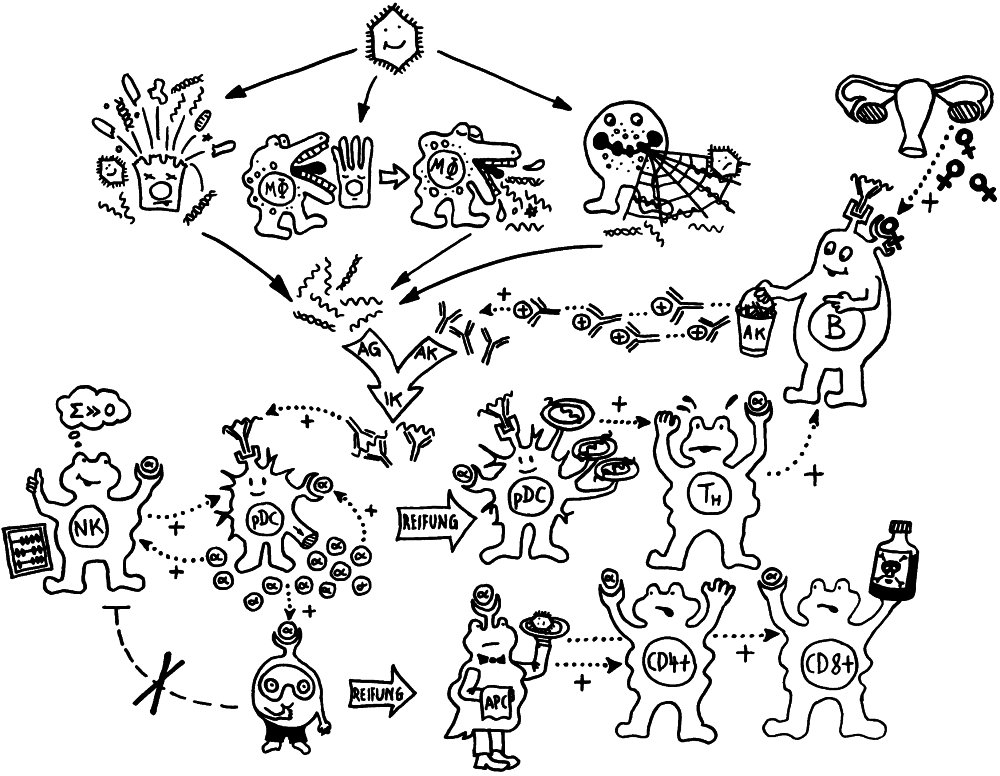
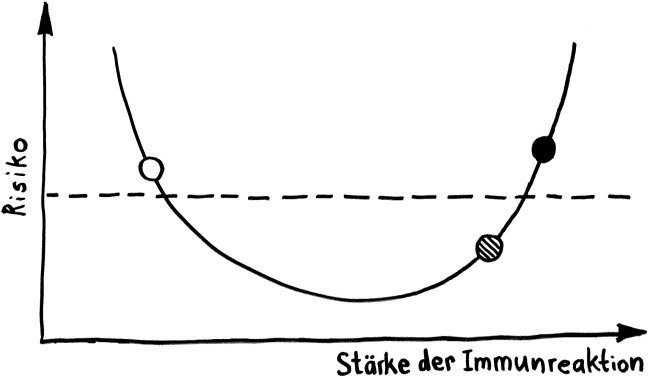
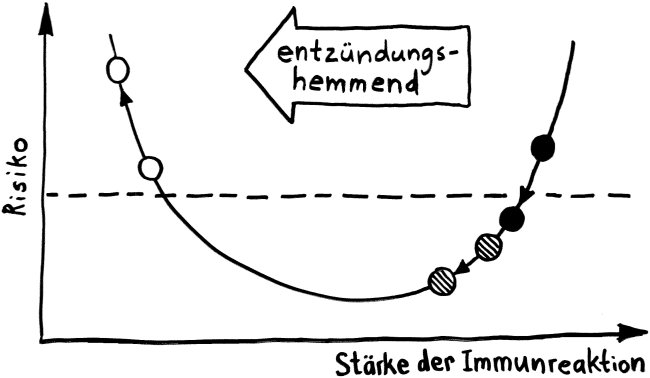
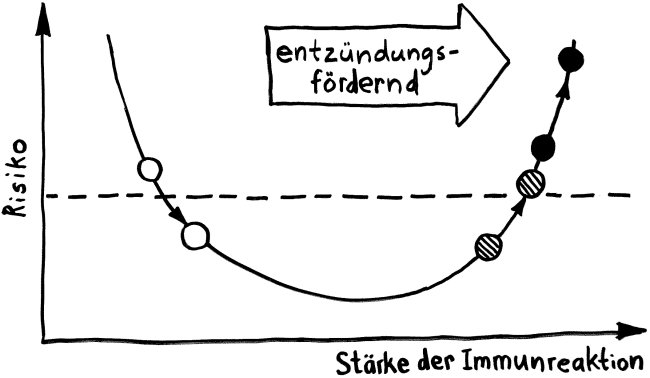
Schnelle Notizen zu 14 kürzlich gelesenen Artikeln – nicht allgemein verständlich aufbereitet, nicht korrekturgelesen und in dieser Form wahrscheinlich nur für mich selbst nützlich. 🙂 Das Ganze wird im letzten Teil des Buches verwurstet, in dem ich die Evolution unseres Immunsytems chronologisch abhandle.
Schnelle Notizen zu 14 kürzlich gelesenen Artikeln – nicht allgemein verständlich aufbereitet, nicht korrekturgelesen und in dieser Form wahrscheinlich nur für mich selbst nützlich. 🙂 Das Ganze wird im letzten Teil des Buches verwurstet, in dem ich die Evolution unseres Immunsytems chronologisch abhandle.
Gibbons A. (2014): Neandertals and moderns made imperfect mates. Science 343, 31.01.2014 (News zu den Arbeiten von Sankararaman et al. 2014, s. u., sowie Vernot & Akey 2014)
Vernot & Akey haben nur moderne Humangenome aus dem 1000 Genomes Project verglichen und daraus Rückschlüsse auf Neandertaler-Einkreuzungen gezogen; Sankararaman et al. haben auch Neandertaler-Genomsequenz einbezogen. Neandertaler haben Spuren in Haut, Nägeln und Haaren (Keratin) hinterlassen; Nachfahren der Hybriden waren weniger fruchtbar als „reine“ moderne Menschen.
In über 60% von 1004 ostasiatischen und europäischen Genomen Neandertaler-Version des Keratinfunktion-Gens. Keratin macht Haut wasserdicht, blockiert Pathogene, macht Haut wärme- und kälteempfindlich -> Anpassung an kältere Habitate?
Neandertaler-Allele, die Risiko für Krankheiten wie Lupus, Morbus Crohn usw. erhöhen, haben Neandertalern vermutlich nicht geschadet, passten aber schlecht zum neuen Kontext im modernen Menschen.
Weitere Neandertaler-Allele -> Hautfarbe.
In allen untersuchten modernen Humangenomen zusammen 20 bzw. 30% des Neandertaler-Genoms wiedergefunden; in einem Individuum stammen 1-3% des Genoms vom Neandertaler. Einkreuzung vor etwa 60.000 Jahren.
Etwa 20 Regionen des Humangenoms enthalten keine Neandertaler-DNA -> negative Selektion wegen Fortpflanzungsnachteilen der Hybriden. Frauen bleiben wegen doppeltem X-Chromosom eher fruchtbar -> Jetzt wird untersucht, ob wir mehr DNA von weiblichen als von männlichen Neandertalern übernommen haben. (Gemeint ist wahrscheinlich das Geschlecht der gemischten Kinder, nicht des reinen Neandertaler-Elternteils – da macht es keinen Unterschied, solange männliche Hybriden mit Neandertaler-X und modernem Y ebenso (un)fruchtbar sind wie männliche Hybriden mit modernem X und Neandertaler-Y.)
Sankararaman S. et al. (2014): The genomic landscape of Neanderthal ancestry in present-day humans. nature, doi:10.1038/nature12961
Vergleich zwischen Neandertaler-Genomen und 1004 modernen Genomen (darunter 176 Yoruba, mutmaßlich Neandertaler-frei) -> Neandertaler-Haplotypen abgeleitet. Regionen mit vielen Neandertaler-Allelen enthalten viele Gene, die Keratinfilamente beeinflussen -> Haut und Haar -> Anpassung moderner Menschen an außerafrikanische Umwelt erleichtert? Große Neandertaler-Allel-freie „Wüsten“ im Humangenom, z. B. auf X-Chromosom, das viele Gene für männliche Fruchtbarkeit enthält; nur teilweise durch geringe Populationsgröße kurz nach Einkreuzung zu erklären -> negative Selektion, evlt. weil Neandertaler-Allele im Genom-Kontext des modernen Menschen Fruchtbarkeit minderten.
Haplotyp-Längen -> Kreuzung vor etwa 2000 Generationen, also 37.000-86.000 Jahren. Neandertaler-Anteil in individuellen Genomen: heute durchschnittlich 1,15% in Europa, 1,38% in Ostasien; kurz nach Einkreuzung über 3% (abgeleitet aus Anteil in „Nicht-Wüsten-Regionen“). Größerer Anteil in Ostasiaten evtl. wegen über lange Zeit kleinerer Populationen als in Europa -> negative Selektion weniger effektiv. Mutmaßlichem Neandertaler-Anteil an einzelnen Genorten: bis zu 62% in ostasiatischen, bis zu 64% in europäischen Populationen. In einigen dieser Regionen Anzeichen für positive Selektion, an an deren negative Selektion.
Aus Neandertalern stammende Allele beeinflussen Risiko für SLE/Lupus, primär biliäre Zirrhose (beides: Transportin-3), Morbus Crohn (Chromosom 10: Zinkfinger-Protein 365, Chromosom 12: Gen unbekannt?), IL-18-Level (Regulator der angeborenen und erworbenen Immunität) , Typ-2-Diabetes, Rauchen und Größe des Blinden Flecks.
Obwohl bei der Einkreuzung nur etwa fünfmal mehr Zeit seit der Aufspaltung zwischen Neandertalern und Vorfahren der modernen Menschen vergangen war als heute seit der Aufspaltung zwischen Europäern und Westafrikanern, war die Fruchtbarkeit der Hybriden wohl wegen Schneeball-Effekten (Dobzhansky-Müller-Inkompatibilitäten) stark reduziert.
Prüfer K. et al. (2014): The complete genome sequence of a Neanderthal from the Altai Mountains. Nature 505, doi:10.1038/nature12886
Hochwertige Genomsequenz einer Neandertaler-Frau aus der Denisova-Höhle in Altai-Gebirge, Sibirien – gewonnen aus einem Zehenknochen aus einer etwa 50.000 Jahre alten Schicht. In derselben Höhle, aber in einer etwas jüngeren Schicht wurde auch der Fingerknochen gefunden, aus dem die vorläufige Genomsequenz des Denisova-Menschen ermittelt wurde. Vergleich mehrerer Neandertaler-Genome (auch aus dem Kaukasus und Kroatien, s. Karte Abb. 1), des Denisova-Menschen-Genoms und 25 moderner Humangenome -> Modell der Einkreuzungsereignisse zwischen modernem Menschen, Denisova, Neandertaler und einem unbekannten Hominiden (Abb. 8). Weiterlesen →