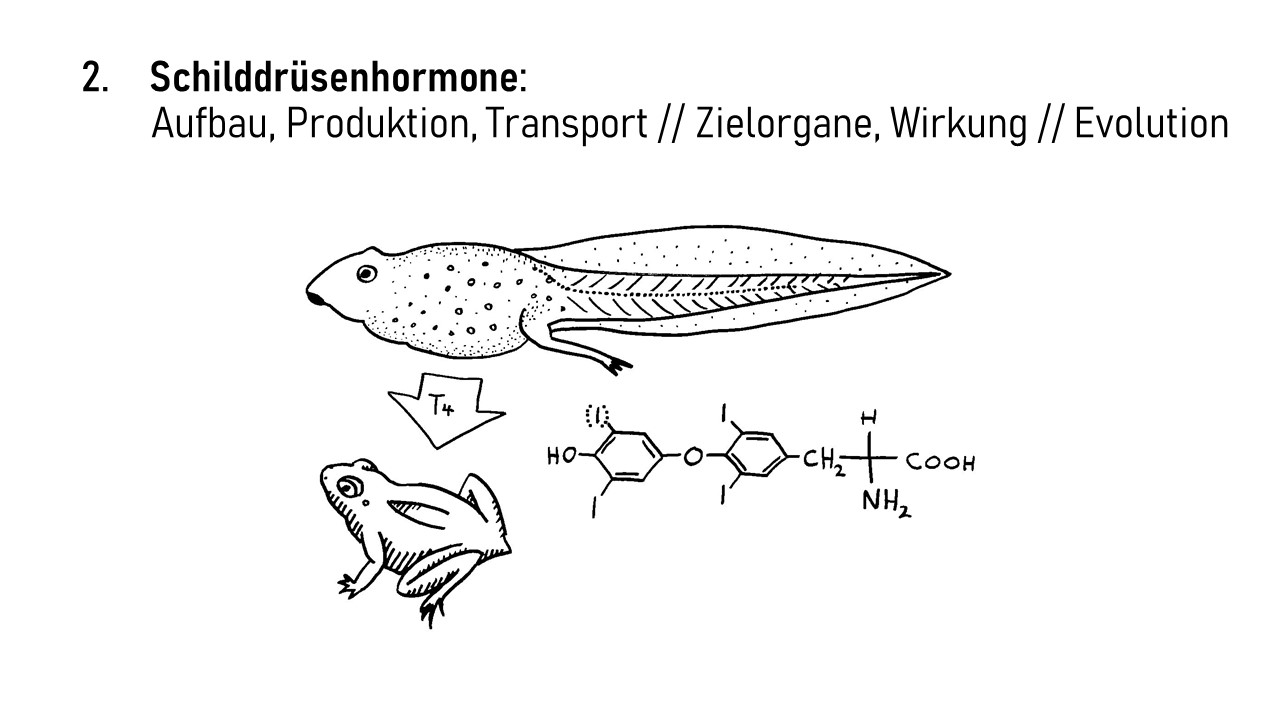
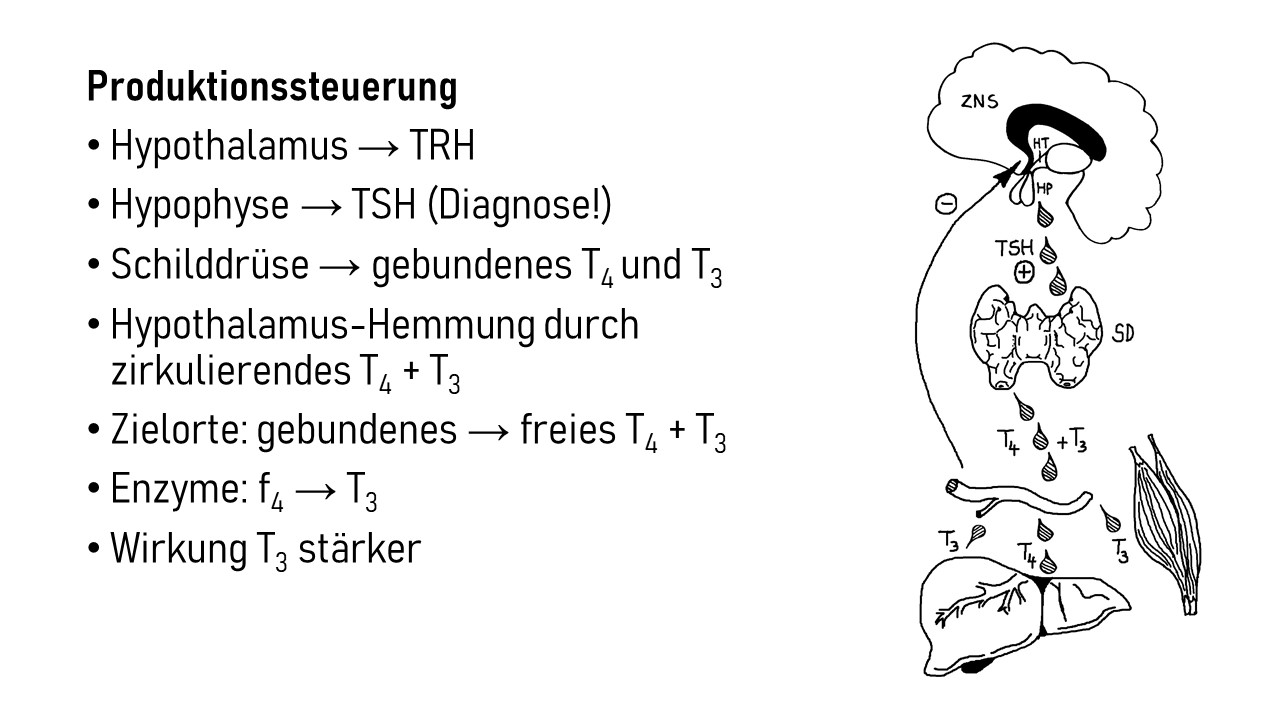
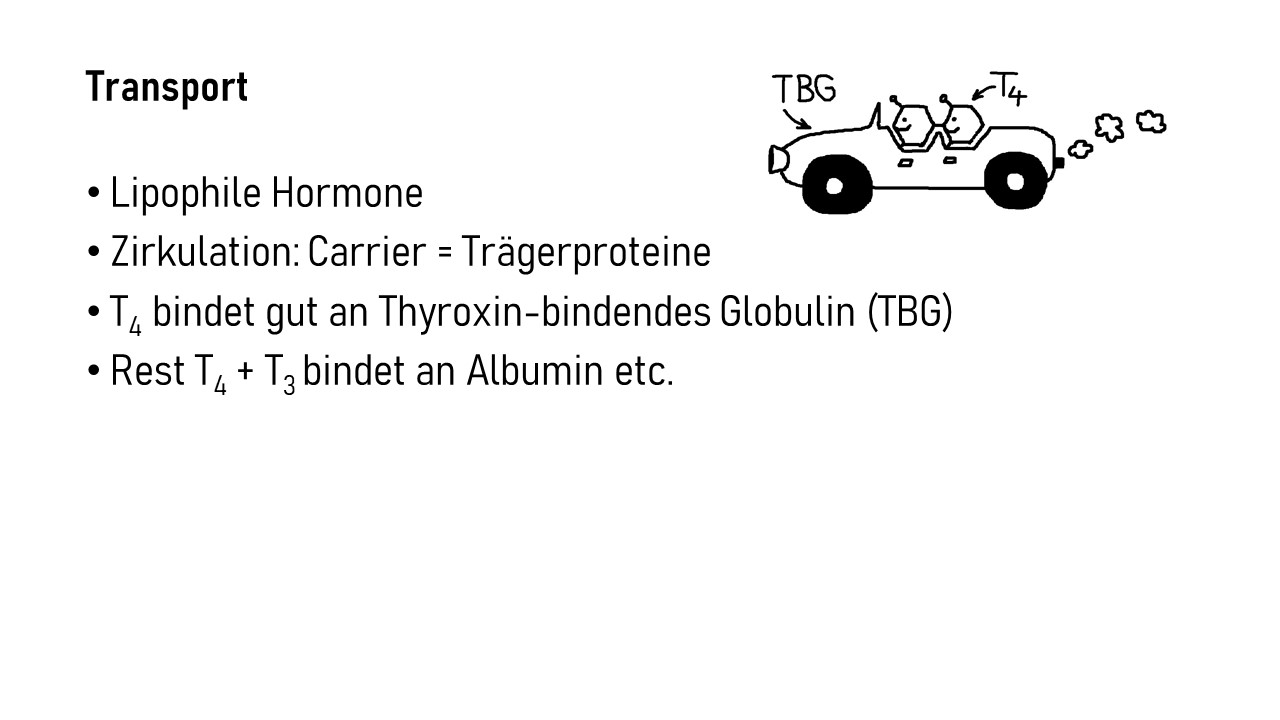
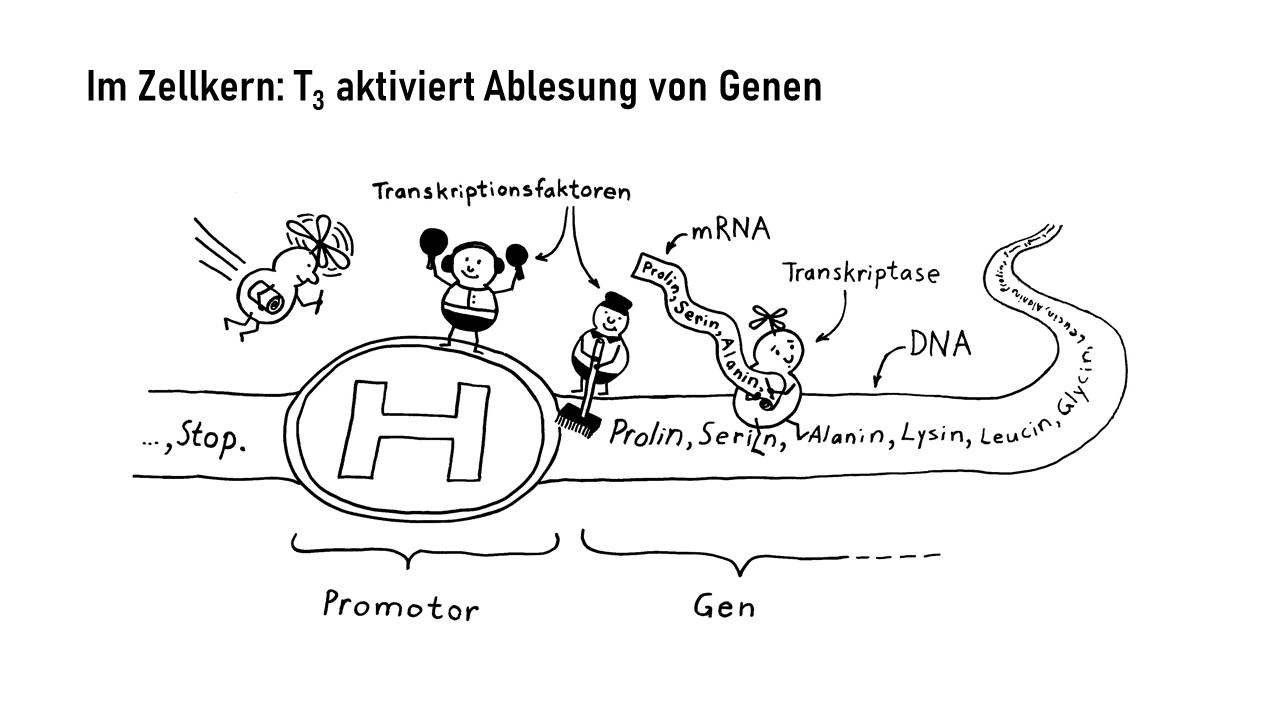
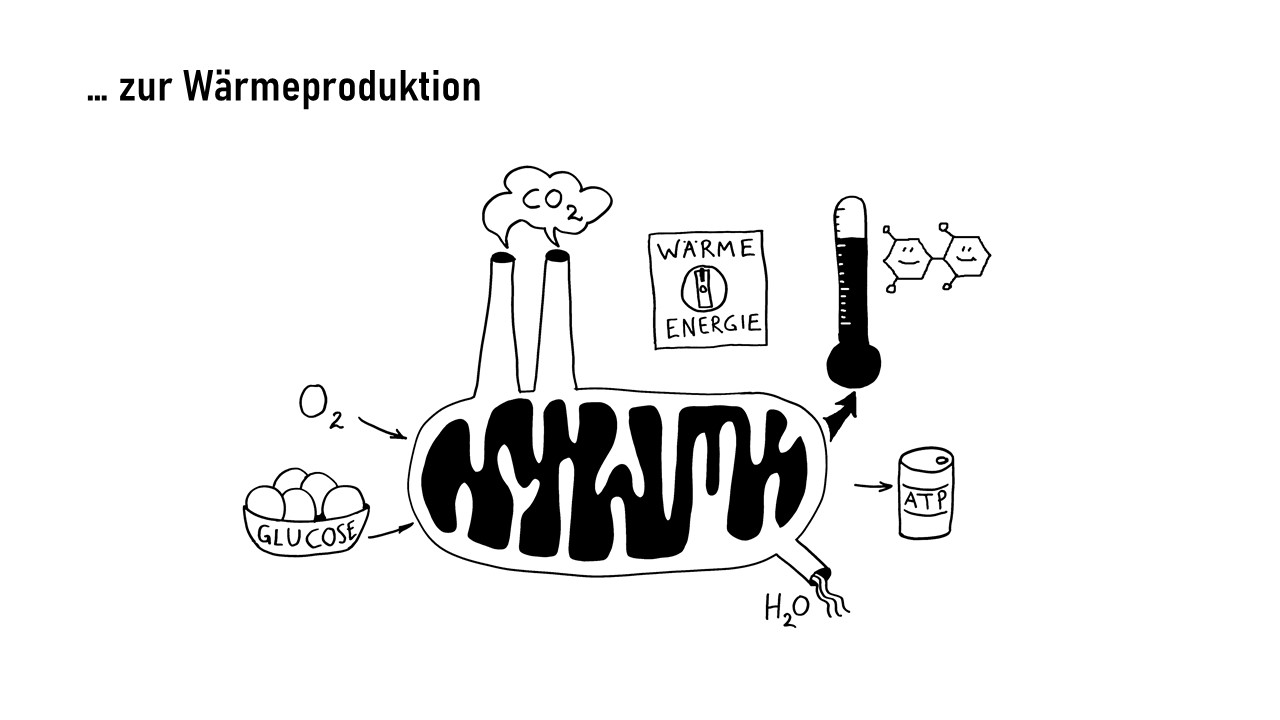
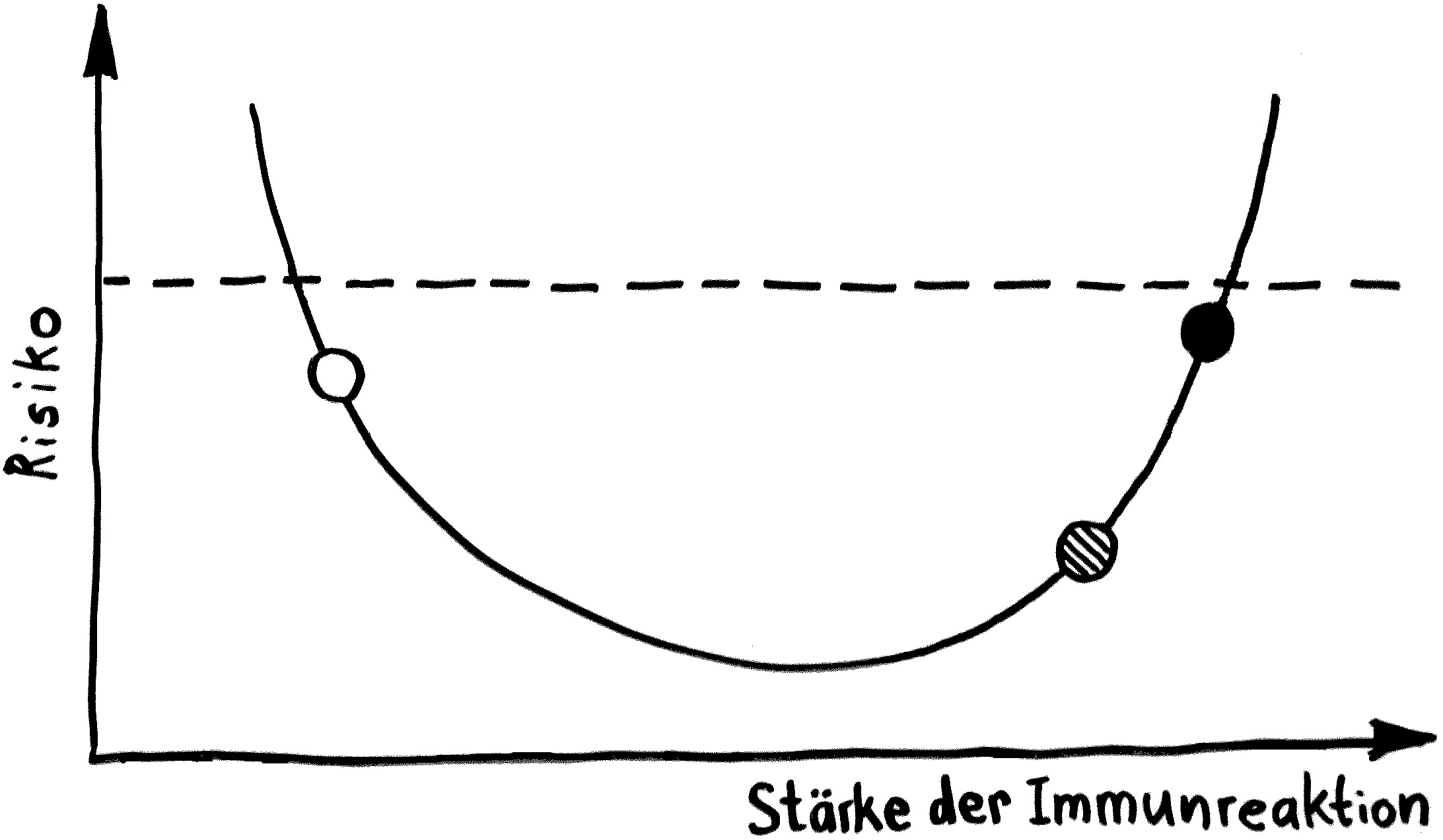
Interleukin-1 (IL-1) ist ein proinflammatorisches Cytokin, also ein Botenstoff, der Entzündungsreaktionen auslöst oder fördert. Er wird in allen möglichen Zellen unseres Körpers produziert und liegt zunächst als inaktive Vorform im Zytoplasma, also im Zellinneren vor. Erst wenn die Zelle ein Alarmsignal an ihre Umgebung aussenden will, etwa weil sie eine Verletzung oder Infektion spürt, die eine Immunreaktion erfordert, bearbeitet sie das Protein so, dass es aus der Zelle austreten kann. Im Zwischenzellraum kann IL-1 dann an einen IL-1-Rezeptor auf der Außenseite einer anderen Zelle binden, etwa einer Immunzelle, die dadurch aktiviert wird und eine Signalkette auslöst, die zu Entzündungsreaktionen beiträgt. So können zum Beispiel weitere proinflammatorische Interleukin-Gene im Zellkern der Immunzelle abgelesen werden, oder die Immunzelle schüttet bereits produzierte Substanzen aus, die zur Anschwellung und Erwärmung des Gewebes, Gefäßweitung, Anlockung weiterer Immunzellen usw. führen.
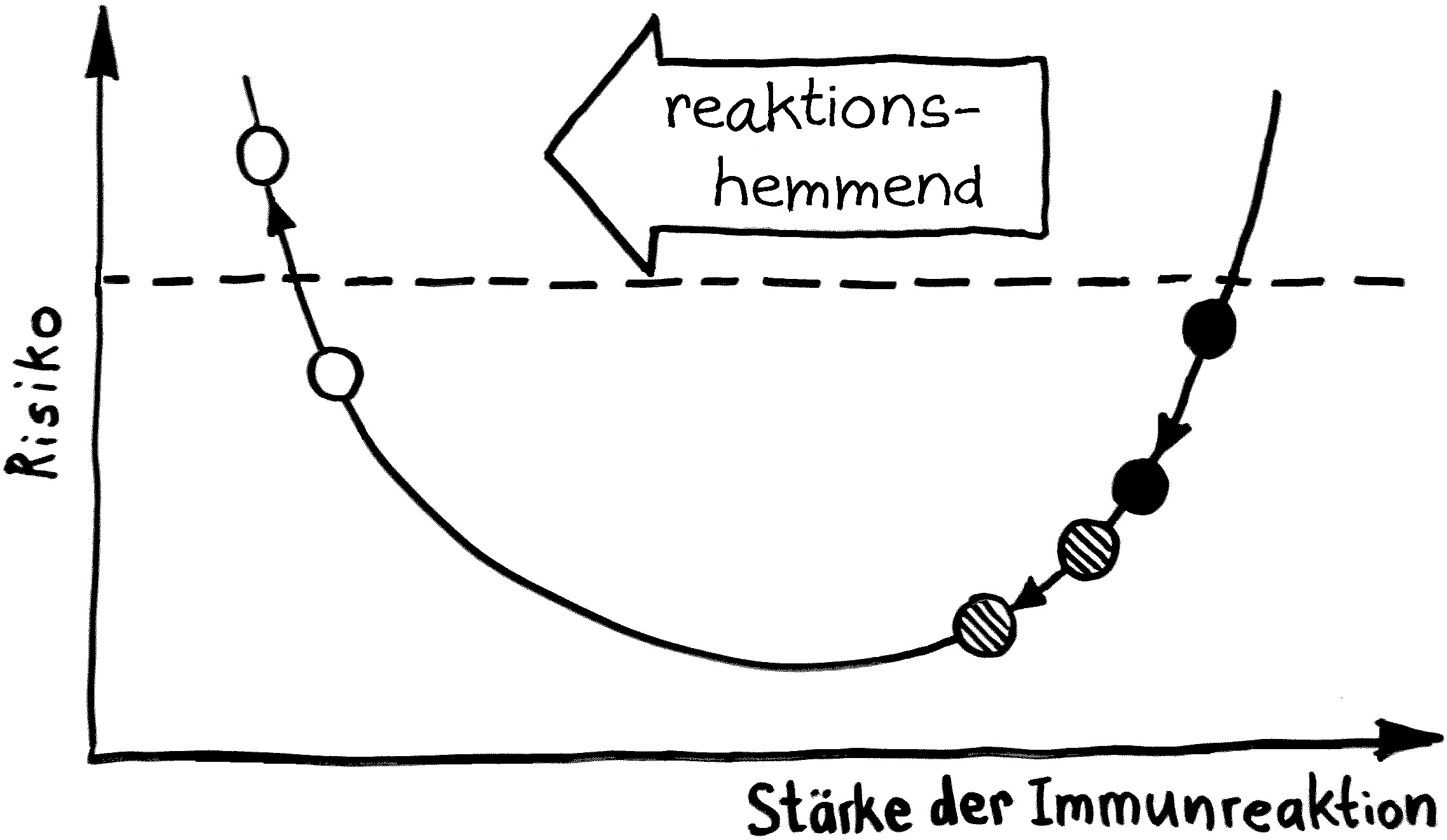
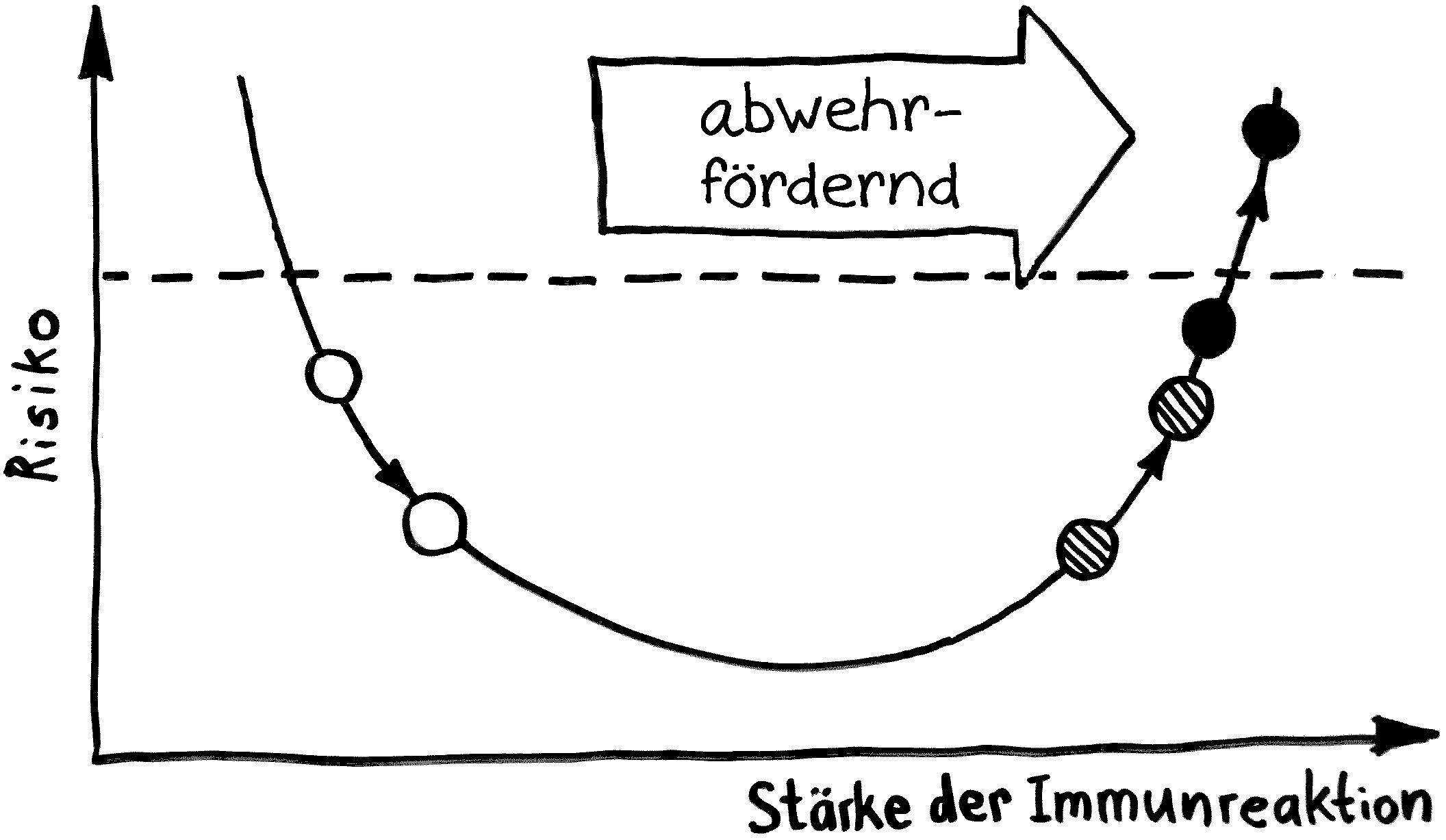
Das darf natürlich nicht ständig passieren, sonst würde jede kleine Störung und jeder Irrtum der ersten Zelle in dieser Reaktionskette zu Entzündungen führen, die dann womöglich einen Teufelskreis auslösen, wie es bei autoinflammatorischen Erkrankungen und Autoimmunerkrankungen der Fall ist. Daher gibt es einen Antagonisten: einen Botenstoff, der dem Interleukin-1 ähnelt und ebenfalls an den IL-1-Rezeptor binden kann, dort aber keine Signalkette auslöst, sondern den Rezeptor blockiert. Dieser IL-1-Rezeptorantagonist (IL-1RA) gehört ebenfalls zur Grundausstattung fast aller unserer Zellen und legt im Normalfall den Großteil der Rezeptoren lahm. Erst wenn wirklich viel IL-1 im Zellzwischenraum unterwegs ist, weil zum Beispiel gleich mehrere Zellen in der Umgebung kräftig Alarm geben und IL-1 ausschütten, verdrängt das proinflammatorische IL-1 den antiinflammatorischen Antagonisten IL-1RA von den Rezeptor-Bindungsstellen.