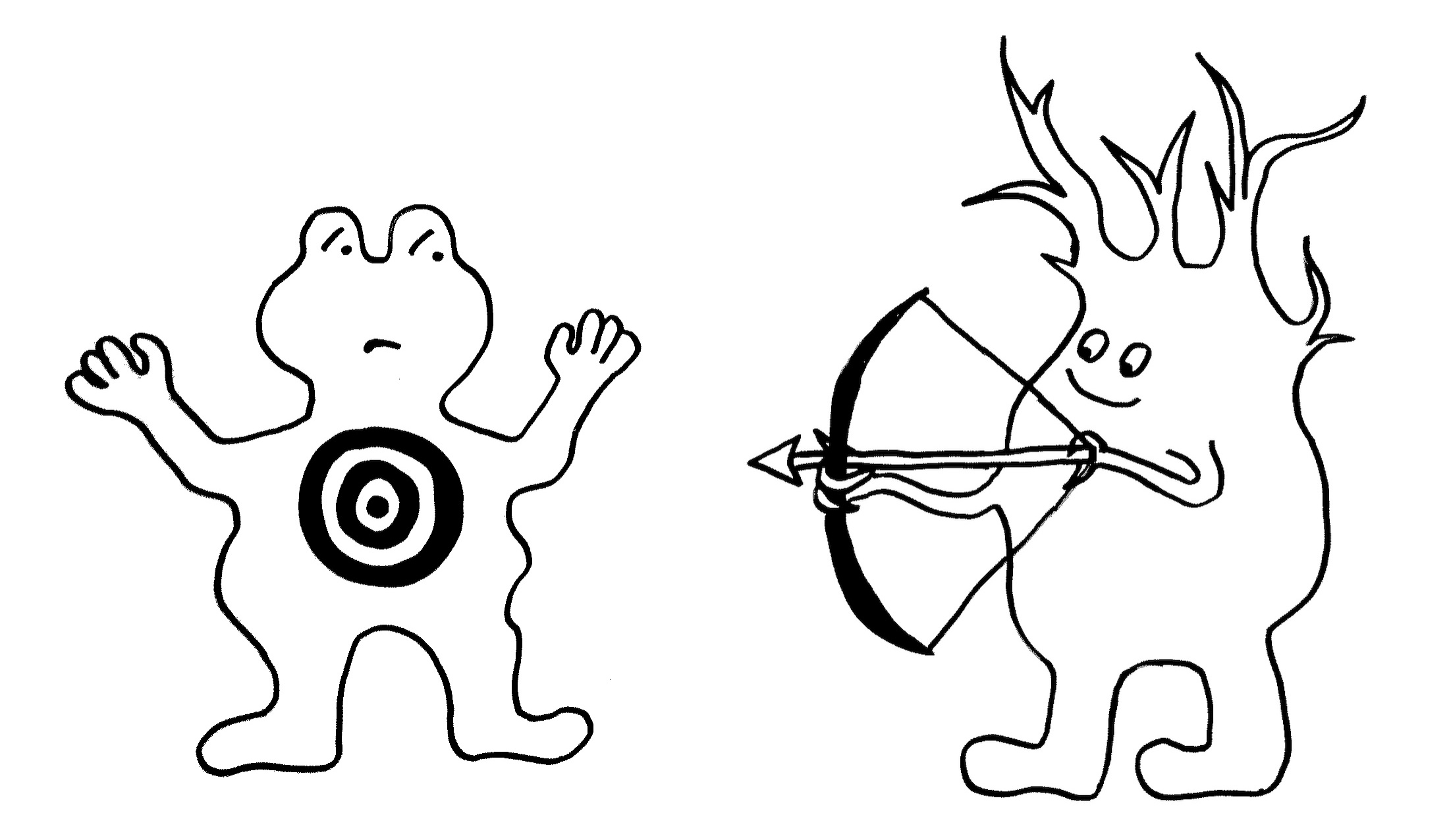
Fas ist ein Rezeptor in der Membran etlicher Zelltypen im menschlichen Körper. FasL ist sein Ligand; auch er ist in der Zellmembran angesiedelt. Bindet FasL (hier: Pfeil und Bogen) an Fas (hier: Zielscheibe), so löst Fas in seiner Zelle eine Apoptose aus, einen kontrollierten Zelltod. Dieser Regulierungsmechanismus kommt in unserem Körper in mehreren Situationen zum Einsatz:
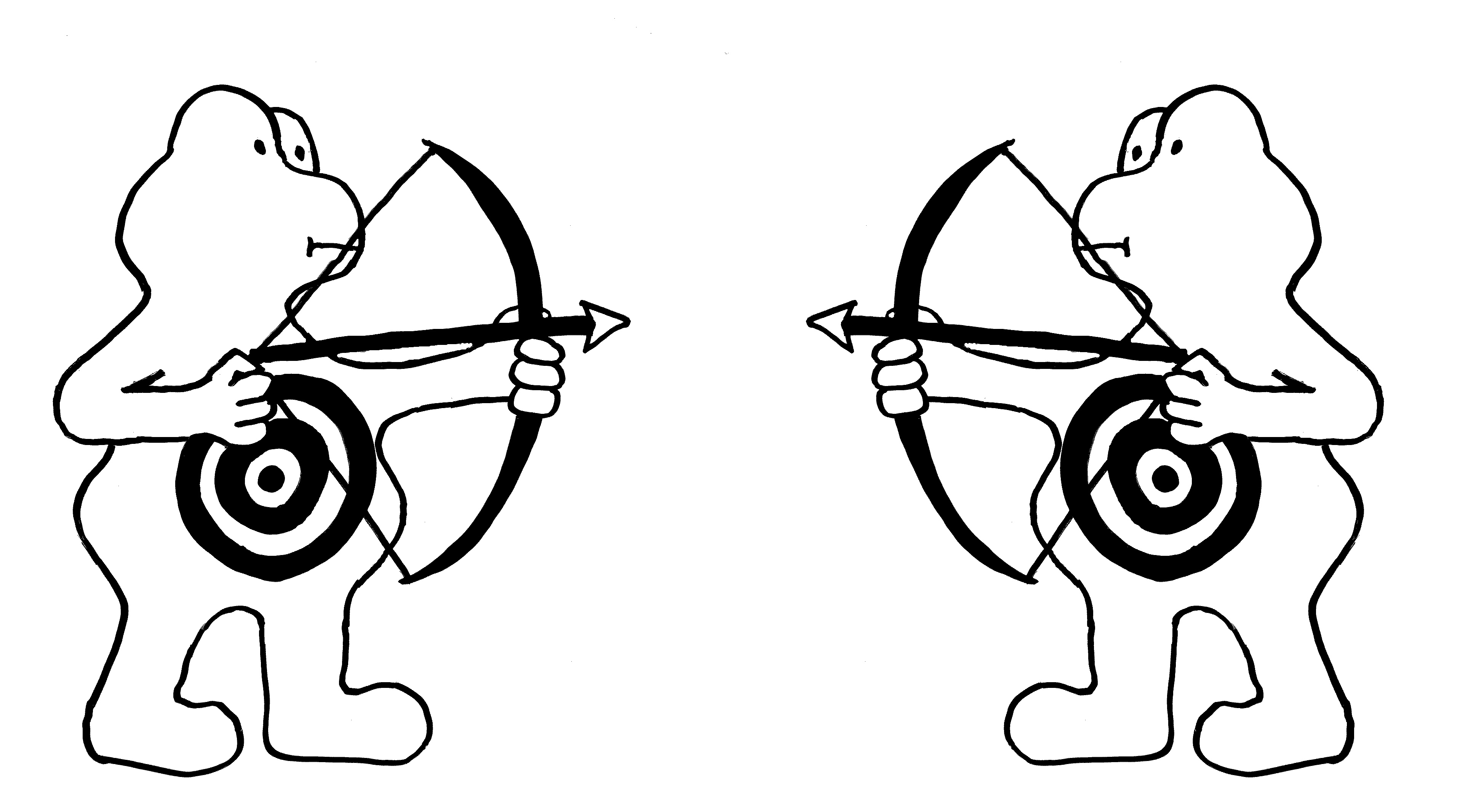
Oben: Zytotoxische T-Zellen (links) bringen so infizierte oder beschädigte Körperzellen (hier eine Nervenzelle, rechts) zum Absterben.
Mitte: In den sogenannten immunologisch privilegierten Orten, beispielsweise im Gehirn, läuft es umgekehrt: Die Zellen dort exprimieren selbst so viel FasL, dass eindringende T-Zellen (deren Membranen sowohl Fas als auch FasL enthalten) sterben, bevor sie eine Abwehrmaßnahme durchführen können.
Unten: Gegen Ende einer Immunreaktion muss die Zahl der Immunzellen im Körper stark reduziert werden (sog. Kontraktion des Immunsystems). Dazu begehen die Immunzellen Brudermord: Da sie sowohl Fas als auch FasL exprimieren, können sie sich gegenseitig ausschalten.
Versagt dieser Kontrollmechanismus, kann es zu Autoimmunerkrankungen kommen.