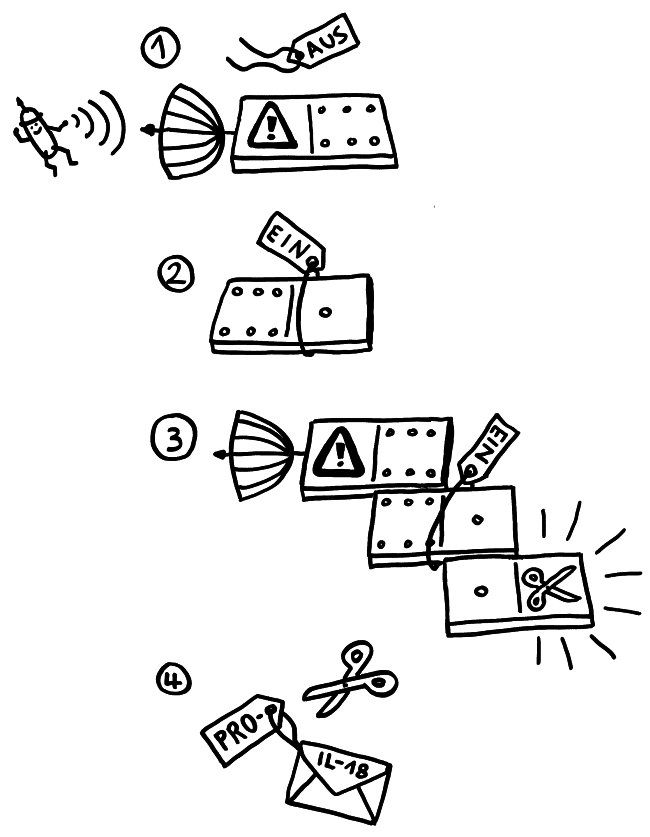
Zeit für einen Werkstattbericht: Manche Abläufe oder Objekte im Immunsystem sind höllisch schwer zu visualisieren. Oft platzt nach tagelanger vermeintlicher Stagnation ein Knoten, und ich wache mit einer Bildidee auf. Aber diese verflixten Inflammasomen – die Proteinenkomplexe in Zellen, die Pyroptose begehen und dabei Entzündungssignale an ihre Umgebung aussenden – sperren sich hartnäckig. Das liegt vor allem an den unterschiedlichen Beschreibungen und Abbildungen in der Fachliteratur: Sie schildern und zeigen ja „nur“ Modelle, die auf dem Kenntnisstand und den Vorstellungen der jeweiligen Autoren vom Aufbau und der Funktionsweise dieser Gebilde beruhen.
Regenschirme, Mühlräder, Raumschiffe
Für diese Funktionsweise sind der dreidimensionale Aufbau und die Dynamik der beteiligten Proteine von entscheidender Bedeutung; beides lässt sich in einfachen zweidimensionalen Schemazeichnungen schlecht einfangen. Hinzu kommt, dass es zahlreiche unterschiedlich aufgebaute Inflammasomen gibt, die jeweils durch andere Signale aktiviert werden. Ich konzentriere mich im Folgenden auf das sogenannte NLRP3-Inflammasom, das am besten erforscht ist. Sein Zusammenbau und seine Aktivität können durch zahlreiche Reize ausgelöst werden, und es scheint bei etlichen chronischen Entzündungen und Autoimmunerkrankungen eine Rolle zu spielen.
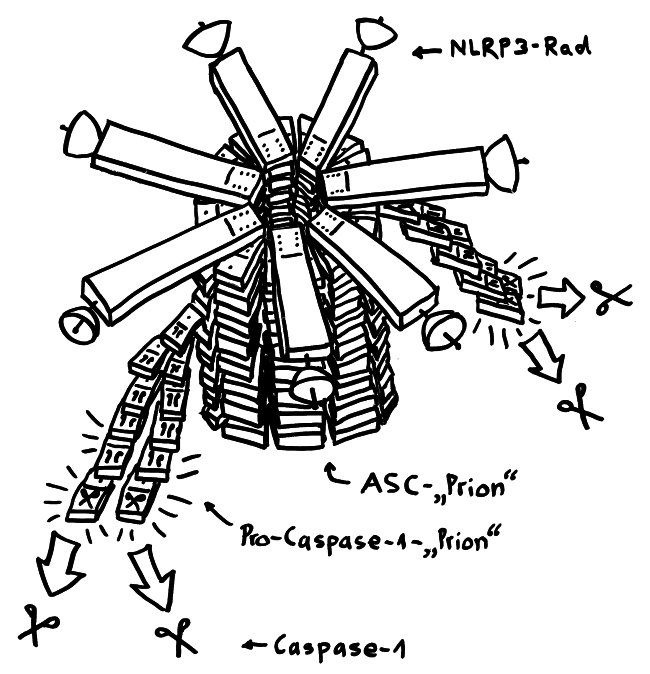
Von oben sieht dieser Proteinkomplex wie ein Regenschirm oder ein Rad mit sechs, sieben oder acht Speichen aus. Von der Seite oder vielmehr im Längsschnitt betrachtet, hat er etwas von einem Raumschiff der Sternenflotte. Er besteht aus drei Baustein-Typen:
- dem namensgebenden Protein NLRP3, das wiederum aus drei Funktionsbereichen oder „Domänen“ zusammengesetzt ist (PYD, NACHT, LRR) und den Sensor enthält, der das Inflammasom aktiviert,
- einem Adapterprotein namens ASC mit zwei Domänen (PYD und CARD) sowie
- dem Enzymvorläufer Pro-Caspase 1, ebenfalls mit zwei Domänen (CARD und p20/10).
Wofür all diese Abkürzungen stehen, das erspare ich uns hier.
Domino
Vielleicht ist es Ihnen aufgefallen: Die Domänen des Adapterproteins, PYD und CARD, tauchen in den beiden anderen Proteinen ebenfalls auf. Das ist kein Zufall, sondern Voraussetzung für den raschen Zusammenbau der Inflammasomen aus den Bausteinen, die in der Zelle bereitliegen und sich mit anderen Bausteinen zusammenlagern können, die dieselben Domänen aufweisen – so, wie man Dominosteine aneinander legt. Im Zytoplasma sind sie normalerweise inaktiv: Sie verbinden sich erst, wenn die Zelle den Weg zur Pyroptose, zum entzündlichen kontrollierten Zelltod einschlägt – sobald sie also Alarmsignale aus Pathogenen (PAMPs) oder aus beschädigten Zellstrukturen (DAMPs) empfängt.
Dieses regulierte Absterben ist Teil der bereits erläuterten angeborenen Immunreaktion, mit der die Ausbreitung von Krankheitserregern eingedämmt, gefährliche Zellüberreste aus dem Gewebe entfernt und Schäden repariert werden. Die Mechanismen sind evolutionär alt und erfordern keine Mithilfe der antigenspezifischen erworbenen Abwehr, also der B- und T-Zellen. Die Gefahrensignale sind nämlich immer dieselben, zum Beispiel reaktive Sauerstoffverbindungen (ROS) aus den eigenen Mitochondrien, Komponenten aus Organellen, die im Zytoplasma eigentlich nichts zu suchen haben und auf eine Beschädigung hinweisen, ein plötzlicher Verlust von Kalzium im Zytoplasma – oder eben typische Bakterienstoffe wie Lipopolysaccharide (LPS). Das eine Ende des Proteins NLRP3 besteht aus dem Rezeptor LRR, der diese Alarmzeichen direkt oder indirekt – über ein zwischengeschaltetes zellinternes Signal – erspürt.
Sobald das passiert, wird zum einen im Zellkern die Produktion von weiteren Inflammasom-Bausteinen hochgefahren. Zum anderen aber lagern sich die bereits fertigen Bausteine zusammen, denn die Zelle muss sofort auf die Gefahr reagieren; die Herstellung von Proteinen dauert ja eine Weile. Am NLRP3 hängt normalerweise ein Label, das das Protein inaktiv hält. Dieses „Schildchen“, Ubiquitin, wird bei einer Aktivierung des Rezeptors LRR abgeknipst, und NLRP3 wird aktiv. Auch das Adapterprotein ASC wird (in diesem Fall durch das Anhängen zweier Label, nämlich Ubiquitin und Phosphatgruppen) in Aktionsbereitschaft versetzt. Nun lagern sich mehrere der langgestreckten NLRP3-Proteine mit ihrem PYR-Ende zusammen, sodass sie wie die Speichen eines Rads wirken.
Prionen-ähnliche Proteinkristalle
An die PYR-Ansammlung an der Radnabe lagern sich anschließend seitlich etliche ASC-Proteine an, und zwar ebenfalls mithilfe ihrer PYR-Einheiten. Sie bilden gewissermaßen eine Radachse, die wie ein Kristall weiterwächst. In der Literatur ist von einem Prionen-artigen Gebilde die Rede. Prionen verbinden wir mit tödlichen Gehirnerkrankungen wie dem Rinderwahn (BSE), der Scrapie beim Schaf und der Creutzfeld-Jakob-Krankheit beim Menschen. Aber auch normale, lebensnotwendige Zellstrukturen wie das Zytoskelett oder eben Teile der Inflammasomen sind im Grunde Proteinkristalle mit einem regelmäßigen Aufbau.
Da sich in der ASC-Achse die PYR-Einheiten innen zusammenlagern, ragen die CARD-Domänen des ASC alle nach außen. An diese docken nun die gleichartigen CARD-Einheiten der Pro-Caspase-1-Bausteine an: wieder das Domino-Prinzip. Das löst die Bildung weiterer Prionen-ähnlicher länglicher Gebilde an, die aus lauter Pro-Caspase-Molekülen bestehen und seitlich von der ASC-Achse weg wachsen. Die nunmehr enge Nachbarschaft vieler dieser Enzymeinheiten hebt ihre Selbsthemmung auf: Sie ändern ihre dreidimensionale Gestalt ein wenig, lösen sich vom Inflammasom und stehen im Zytoplasma als aktive Caspase-1 zur Verfügung. Wie im Beitrag über die Pyroptose erläutert, verwandelt Caspase-1 unter anderem die Vorformen der entzündungsfördernden Zytokine IL-1β und IL-18 in ihre aktiven Formen, die aus der sterbenden Zelle ausgeschieden werden und die Nachbarschaft alarmieren.
Nützlicher Ansteckungseffekt
Offenbar gibt es neben diesen Zytokinen noch einen weiteren Ausbreitungsweg für eine aufkeimende Entzündung. Die sterbende Zelle setzt im Zuge ihrer Pyroptose größere Bruchstücke aus den Prionen-artigen ASC-Achsen der Inflammasomen frei. Diese bereits aktivierten ASC-Kristalle können offenbar auch außerhalb von Zellen Pro-IL-1β zu aktivem IL-1β umwandeln. Außerdem werden sie von Makrophagen vertilgt, die – durch die Zytokine und Gefahrensignale aus der sterbenden Zelle angelockt – den Zellmüll beseitigen. In den Makrophagen können die Enzymkomplexe weiterarbeiten und Pro-Caspase-1 zu aktiver Caspase-1 umbauen. Durch diese Kettenreaktion kann sich eine Entzündung, die von einigen wenigen Zellen ausgeht, sehr schnell ausbreiten.
Inflammasomen bei Autoimmunerkrankungen: Rolle ungeklärt
Wie viele andere nützliche Strukturen im Immunsystem kann auch das Inflammasom an Autoimmunerkrankungen, chronischen Entzündungen und anderen Krankheiten wie Alzheimer-Demenz, Parkinson oder Diabetes mitwirken, wenn es zur falschen Zeit, am falschen Ort, zu stark oder zu schwach aktiv wird. Das wiederum kann durch kleine Varianten (sogenannte Einzelnukleotid-Polymorphismen oder single nucleotide polymorphisms, kurz SNPs) in den vielen an seinem Aufbau und seiner Arbeit beteiligten Genen passieren. Solche SNPs in den Genen für Inflammasom-Komponenten, die in einzelnen Ethnien das Risiko einer Autoimmunerkrankung erhöhen, den Verlauf der Erkrankung verschlimmern oder die Wirksamkeit einer Therapie dagegen verringern könnten, sind bislang für Vitiligo, Lupus, rheumatoide Arthritis, juvenile idiopathische Arthritis, systemische Sklerose, Nebennierenrindeninsuffizienz und Psoriasis bekannt.
Bei vielen Autoimmunerkrankungen verschlimmert eine übermäßige IL-1β und IL-18-Ausschüttung den Verlauf, was vermuten lässt, dass bestimmte Zellen der Betroffenen überaktive Inflammasomen beherbergen. Allerdings ist ein kausaler Zusammenhang mit genetischen Veränderungen in den Inflammasom-Genen noch für keine dieser Krankheiten wirklich belegt, und Studien an unterschiedlichen Tiermodellen und an Menschen liefern oft widersprüchliche Ergebnisse. Die Inflammasomen sind eben nur ein Rädchen im hochkomplexen Regulierungssystem, das externe und köpereigene Gefahrensignale, Zelltod und Zellrettung, Organschädigung und Gewebsreparatur miteinander verbindet.
(Zeichnungen im nächsten Beitrag)
Literatur
H. Guo et al. (2105): Inflammasomes: mechanism of action, role in disease, and therapeutics
C.-A. Yang, B.-L. Chang (2015): Inflammasomes and human autoimmunity: A comprehensive review

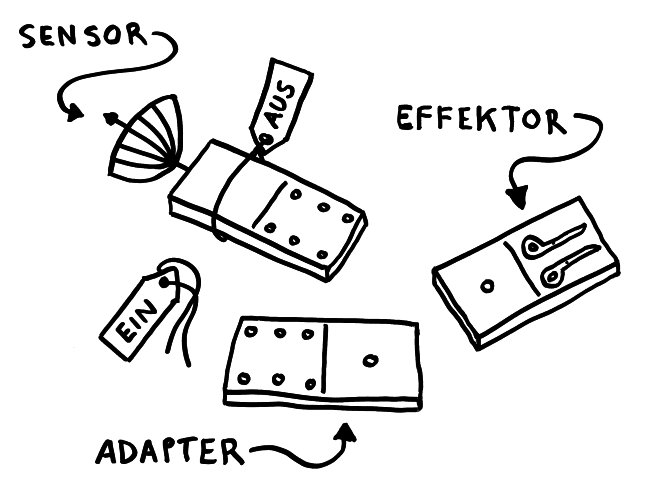 Solange sie einzeln im Zytoplasma der Zelle herumliegen, sind die Bausteine inaktiv – der Sensor, weil er durch ein Protein-Anhängsel inhibiert wird, der Adapter, weil ihm zwei für die Aktivität nötige Anhängsel fehlen, und der Effektor, weil er in dieser Gestalt nicht als Enzym arbeiten kann. Das ändert sich, sobald der Sensor einen Reiz empfängt, etwa ein Bakterien-typisches Alarmsignal:
Solange sie einzeln im Zytoplasma der Zelle herumliegen, sind die Bausteine inaktiv – der Sensor, weil er durch ein Protein-Anhängsel inhibiert wird, der Adapter, weil ihm zwei für die Aktivität nötige Anhängsel fehlen, und der Effektor, weil er in dieser Gestalt nicht als Enzym arbeiten kann. Das ändert sich, sobald der Sensor einen Reiz empfängt, etwa ein Bakterien-typisches Alarmsignal: