Charles A. Janeway (1943-2003)
Wer aus dem Stegreif das Immunsystem definieren soll und dabei auf sein Schulwissen oder seither Angelesenes baut, wird wahrscheinlich ungefähr sagen, dass dieses System das Selbst vor Angriffen von außen schützt, indem es auf Fremdes im Körper reagiert und dieses nach Möglichkeit zerstört, isoliert oder ausscheidet – zum Beispiel Krankheitserreger oder Allergene. Als Vertreter dieses traditionellen Verständnisses, das auch als „self-nonself model“ bezeichnet wird, habe ich hier den Immunologen Charles Janeway skizziert, auf den eines der großen Lehrbücher der Immunologie zurückgeht. Er vertrat eine moderne Variante des Konzepts, der zufolge das Immunsystem nicht jedes „nonself“ angreift, sondern nur „infectious nonself“, also Infektionserreger (s. u.).

Polly Matzinger (*1947)
Vor einigen Jahrzehnten stellte Polly Matzinger dieses Konzept infrage, denn es ließ ihres Erachtens zu viele Phänomene außer Acht – etwa Autoimmunstörungen, bei denen das Immunsystem körpereigenes Gewebe bekämpft, oder die heilsame Wirkung dieses Systems bei sogenannten sterilen Entzündungen, etwa Quetschungen, bei denen keine Keime in den Körper eindringen. Sie nannte ihr Konzept „Danger theory“, denn das Immunsystem reagiert ihr zufolge auf Gefahren aller Art, ob diese nun von außen (aus dem „nonself“) kommen oder im Inneren (im „self“) angesiedelt sind.
Lange galten die Konzepte als unverträglich, und die Anhänger beider Schulen bezichtigten die jeweils andere Schule zum Beispiel, „Selbst“ oder „Gefahr“ tautologisch oder zu schwammig zu definieren. Dahinter steckten auch kulturelle Differenzen. So entstand das traditionelle Bild vom Immunsystem wesentlich im Zweiten Weltkrieg, als man viele Brandwunden zu versorgen hatte und bei den Hauttransplantationen regelmäßig Abstoßungsreaktionen beobachtete, die nur dann nicht auftraten, wenn die Haut von einem eineiigen Zwillingsgeschwister stammte – also von einem zweiten Selbst. Die Begrifflichkeit dieser Schule ist entsprechend kriegerisch: Angriff, Eindringlinge, Abwehr und so weiter. Als sich in den 1970er-Jahren das ökologische Denken ausbreitete, suchte man nach Alternativen: nach einem integrativeren Konzept, in dem zwischen dem Selbst und der Umwelt keine Mauern oder Gräben lagen, sondern durchlässige „grüne Grenzen“.

Nach Matzinger reagiert das Immunsystem nicht auf alles, was nicht zum Körper gehört (alles außerhalb des linken Kreises, „nonself“) und auch nicht auf alle Bakterien, Viren usw. (rechter Kreis, „infectious nonself“), sondern auf Gefahren (mittleres Oval, „danger“) – ob nun körpereigene wie Krebszellen oder körperfremde wie Pathogene. Autoimmunerkrankungen wären demnach in derselben Schnittmenge zwischen Selbst und (vermeintliche) Gefahr angesiedelt wie Krebs.
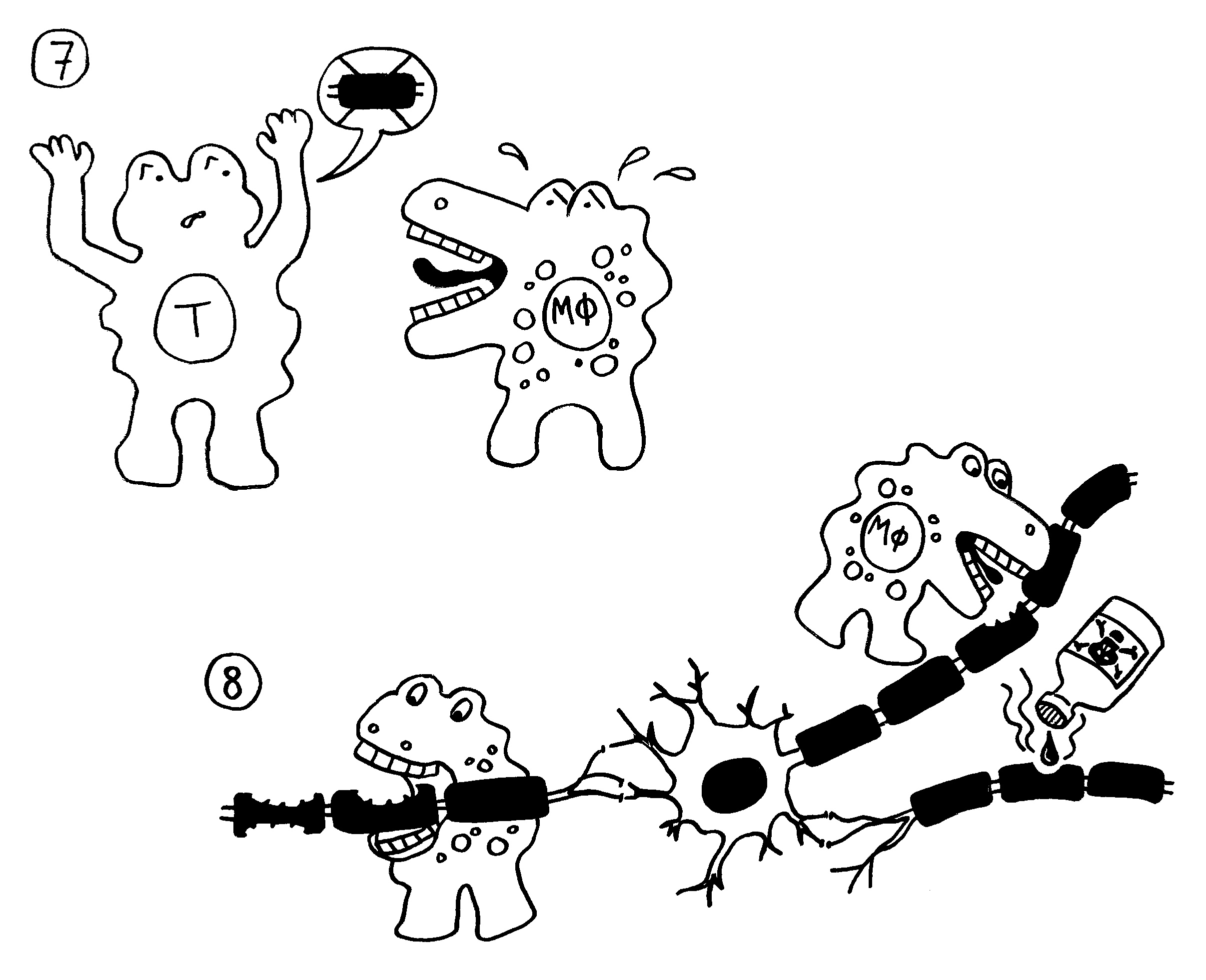
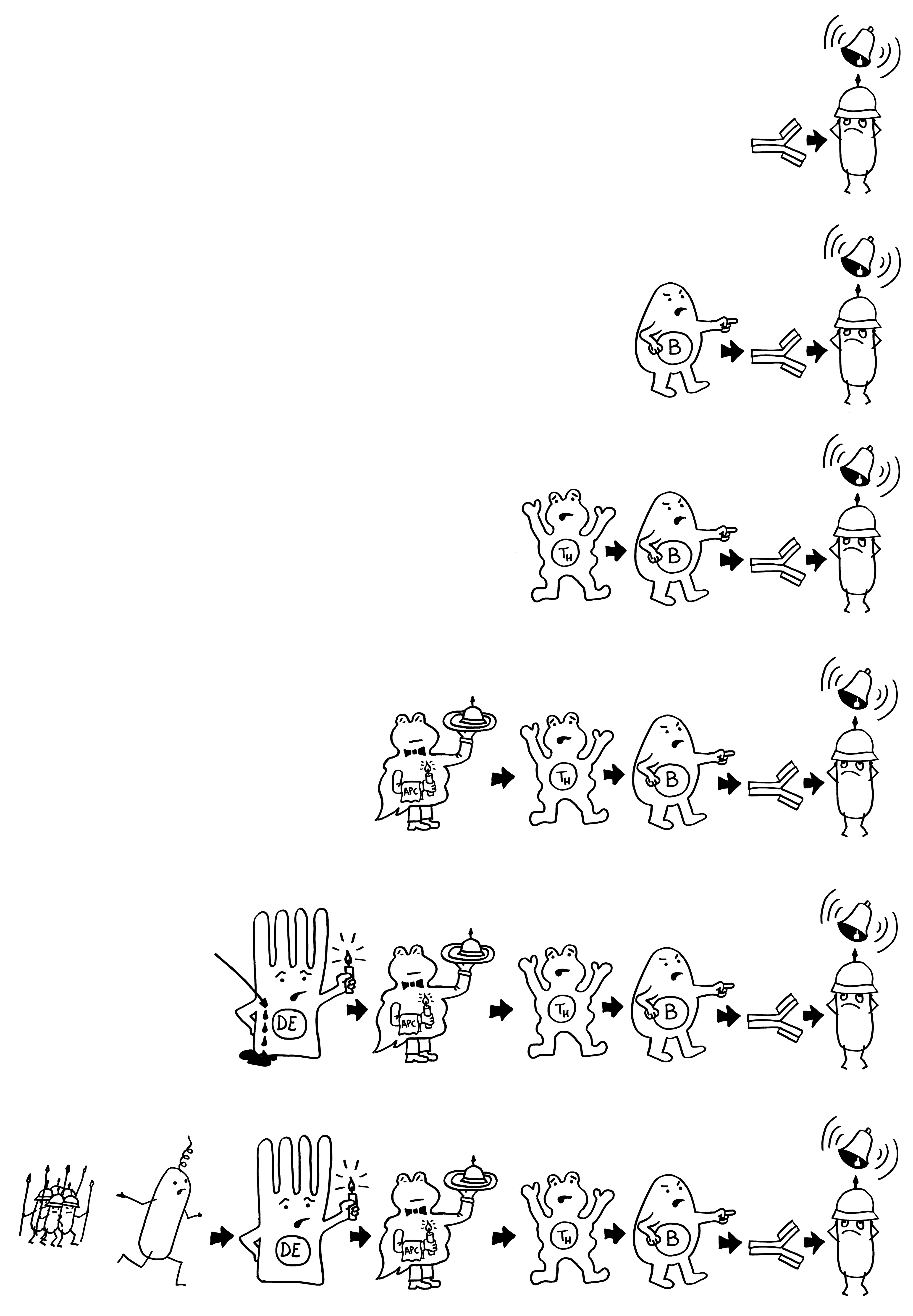
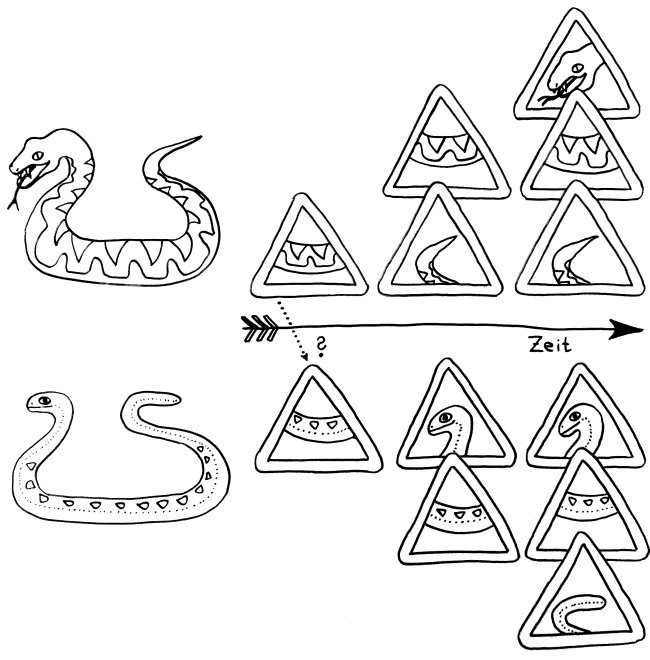
Doch welche Instanzen in unserem Körper entscheiden, wann eine Gefahr vorliegt, und setzen eine angemessene Immunreaktion in Gang? In ihren Essays „The Danger Model in Its Historical Context“ und „The Danger Model: A Renewed Sense of Self“ stellt Matzinger die Entwicklung der immunologischen Modelle und die wiederholte Vorverlagerung dieser Kontrollinstanz dar. Den Anfang macht das ursprüngliche, von Frank Macfarlane Burnet 1959 vorgestellte Self-nonself-Modell, dem zufolge die Lymphozyten (hier eine B-Zelle) aktiviert werden, sobald sie ein fremdes Antigen erkennen. Demnach hätte das Antigen (hier ein Bakterium) die Kontrolle:

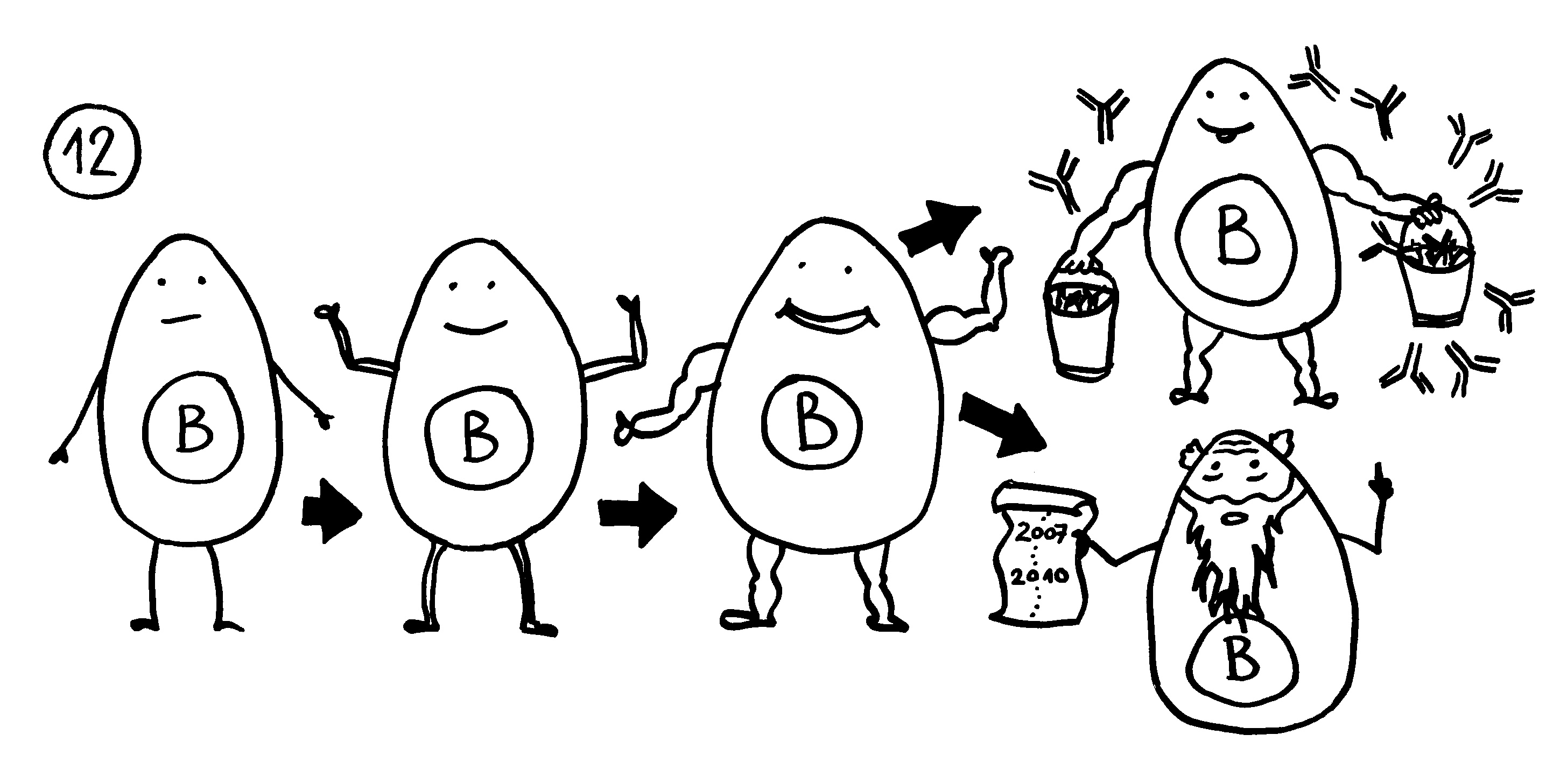
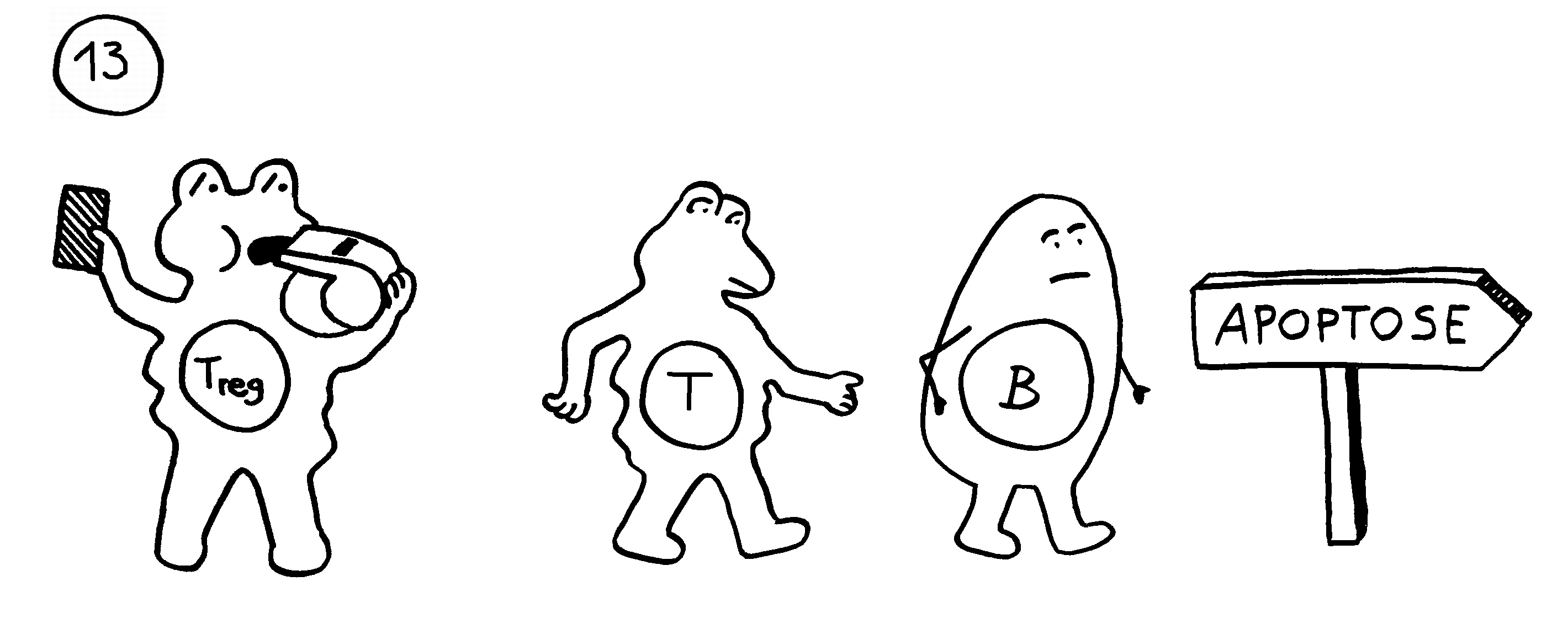
Auf dieselbe Weise sollten auch T-Zellen infizierte Körperzellen erkennen und dann gegen sie vorgehen. 1969 erweiterten P. Bretscher und M. Cohn das Modell um ein zweites Signal, das eine Absicherung gegen zerstörerische Überreaktionen darstellt. Denn man hatte entdeckt, dass einmal aktivierte B-Zellen durch somatische Hypermutation eine Vielzahl leicht unterschiedlicher Rezeptoren hervorbringen, von denen sehr viele an körpereigene Antigene binden und so ständig Autoimmunreaktionen auslösen müssten. Also postulierte man, dass nur solche B-Zellen überleben, die das von ihren Rezeptoren erkannte Antigen einer T-Helferzelle vorzeigen (2) und von dieser die Bestätigung erhalten, dass es sich um ein fremdes Antigen handelt (3). In diesem Modell hat die T-Helferzelle die Kontrolle über die Immunreaktion:
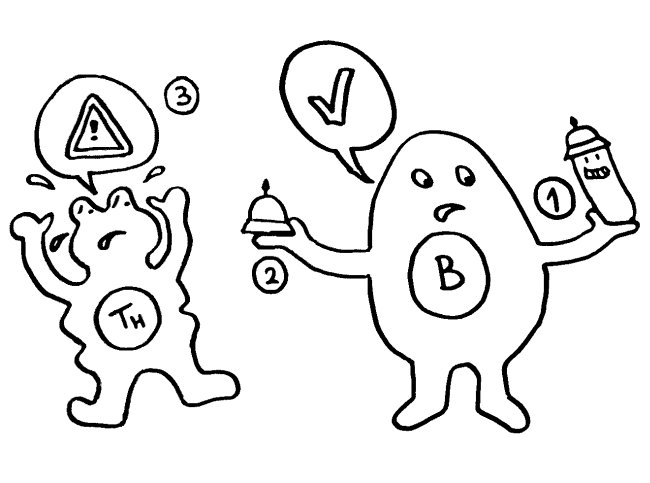
1974 ergänzten K. J. Lafferty und A. J. Cunningham das Tableau um einen weiteren Zelltyp, der heute als antigenpräsentierende Zelle (APC) bekannt ist, und ein weiteres Signal, die sogenannte Kostimulation (3). Denn wie sich gezeigt hatte, sind T-Helferzellen im Grundzustand inaktiv. Wie die B-Zellen brauchen sie, um richtig aufzuwachen und Alarm zu schlagen, mehr als bloß Kontakt zu einer B-Zelle, die ihr ein passendes Antigen vorzeigt. Fehlt die Kostimulation durch eine APC, sterben die T-Helferzellen und in der Folge auch die B-Zellen – oder aber die T-Helferzellen werden tolerant, d. h. sie schlagen nicht Alarm, sondern wirken vielmehr beschwichtigend auf die B-Zellen ein. In diesem Modell steuert also die antigenpräsentierende Zelle die Immunreaktion:
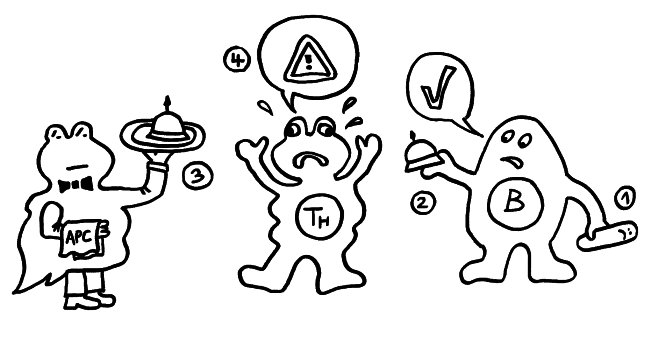
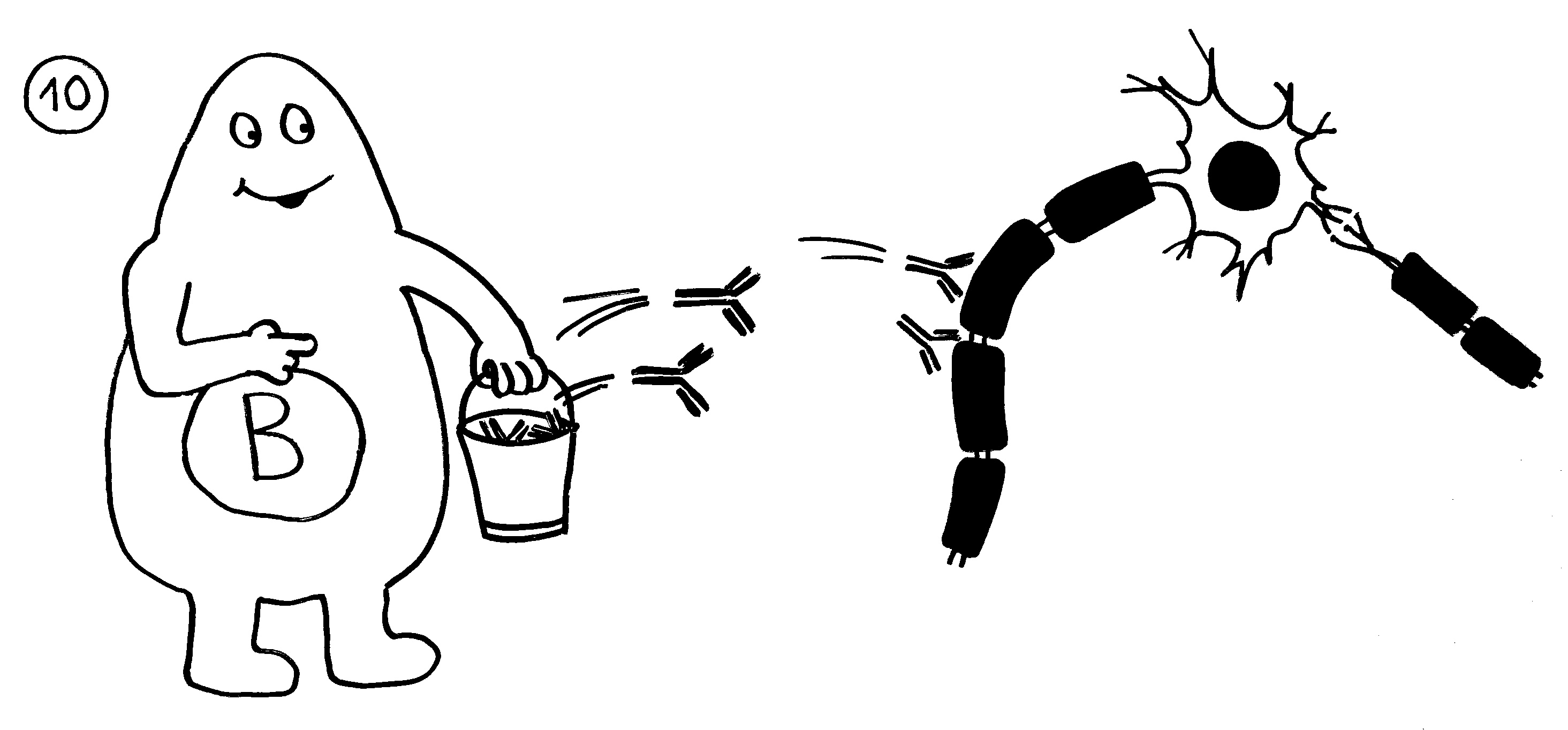
Rätselhaft blieb allerdings, wieso ausgerechnet antigenpräsentierende Zellen – also Makrophagen und dendritische Zellen – diese wichtige Kontrollfunktion ausübern sollten. Schließlich haben sie als Zellen der angeborenen Abwehr keine antigenspezifischen Rezeptoren und können gar nicht zwischen fremden, potenziell gefährlichen Antigenen und irgendwelchen harmlosen körpereigenen Proteinschnipseln usw. unterschieden.1989 schlug der eingangs erwähnte Charles Janeway eine Lösung vor.
Ihm zufolge können die APCs getrost alles präsentieren, was sie finden, ob nun fremd oder körpereigen (1). Die T-Helferzellen werden nur dann aktiv, wenn sie gleichzeitig ein weiteres Signal empfangen (2), das nur entsteht, wenn die APC über bestimmte Rezeptoren, die „pattern recognition receptors“ (PRRs), typische Strukturen erkennen, die eindeutig nicht aus dem eigenen Körper, sondern von evolutionär sehr weit entfernen Organismen stammen müssen. Solche Strukturen oder Muster werden als „pathogen-associated molecular patterns“ (PAMPs) bezeichnet. Ein typisches Beispiel sind Lipopolysaccharide (LPS), die in der äußeren Membran sehr vieler Bakterien vorkommen: In unserem Körper gibt es nichts Ähnliches. Also sind auch die antigenpräsentierenden Zellen nicht ständig aktiv, sondern nur, wenn sie Gefahr wittern. In diesem Modell haben letztlich die PRRs die Kontrolle:
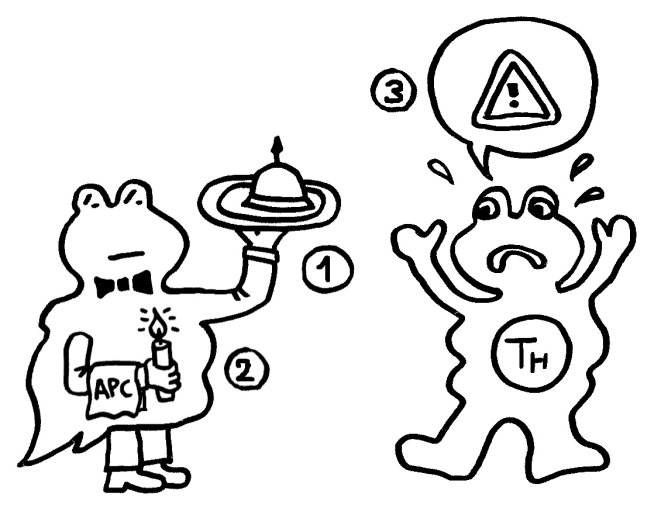
Aber Janeways „infectious nonself“-Modell kann nicht erklären, warum das Immunsystem auch auf Transplantate oder Tumoren reagiert, die ja keine für die PRRs erkennbaren Bestandteile evolutionäre sehr fremder Organismen enthalten. Auch Autoimmunerkrankungen bleiben in diesem Modell rätselhaft, denn meistens sind dabei ebenfalls keine Bakterien, Viren oder Pilze im Spiel.
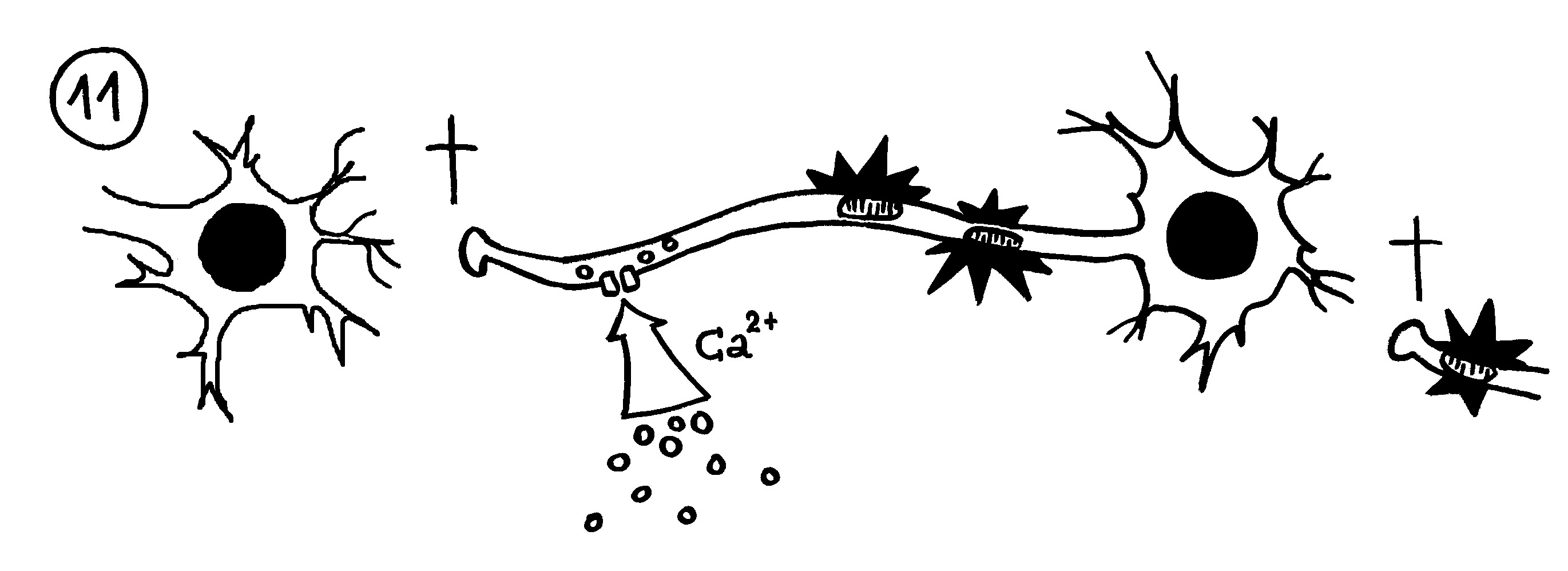
Auftritt Matzinger. Sie schlug 1994 vor, dass die APCs nicht nur durch PAMPS alarmiert werden können, sondern auch durch DAMPs, also „danger-associated molecuar patterns“ oder „damage-associated molecular patterns“. Bei Transplantationen sterben zum Beispiel auf einen Schlag recht viele Zellen des Implantats ab, sobald dieses wieder durchblutet wird. Auch bei Krebs kommt es zu einem massenhaften Zellsterben, und viele Krankheitserreger fallen dem Immunsystem selbst gar nicht auf, verletzen aber zahlreiche Zellen, aus denen dann Substanzen austreten, die normalerweise ausschließlich im Zellinneren zu finden sind – etwa DNA oder RNA. APCs nehmen solche DAMPs zum Teil über dieselben Rezeptoren wahr wie PAMPs, denn beide Substanzklassen sind evolutionär sehr alt und gut konserviert, also nahezu unveränderlich und damit leicht zu erkennen. In diesem Modell hat das verletzte Gewebe die Kontrolle über die Immunreaktion:
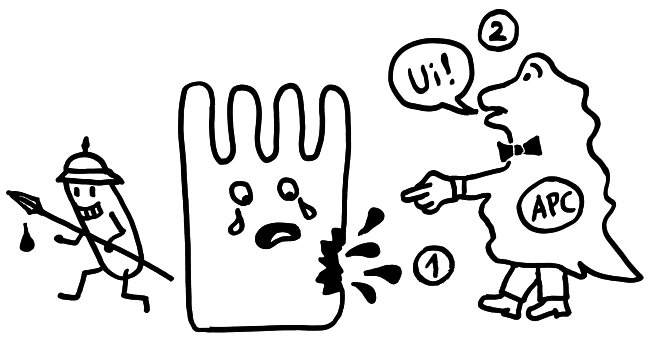
Das Gewebe steuert Matzinger zufolge nicht nur, ob eine Immunreaktion in Gang kommt, sondern teilweise auch, welcher Art sie ist. Denn das jeweilige Gewebe oder Organ weiß selbst am besten, welche Immunreaktion ihm gefährlich werden könnte: Im Auge oder anderen lebenswichtigen Organen werden zurückhaltende Reaktionen benötigt, etwa in Form von Antikörpern in der Tränenflüssigkeit. Andernorts kann die Abwehr energisch zuschlagen, um eine Ausbreitung einer Gefahr zu verhindern.
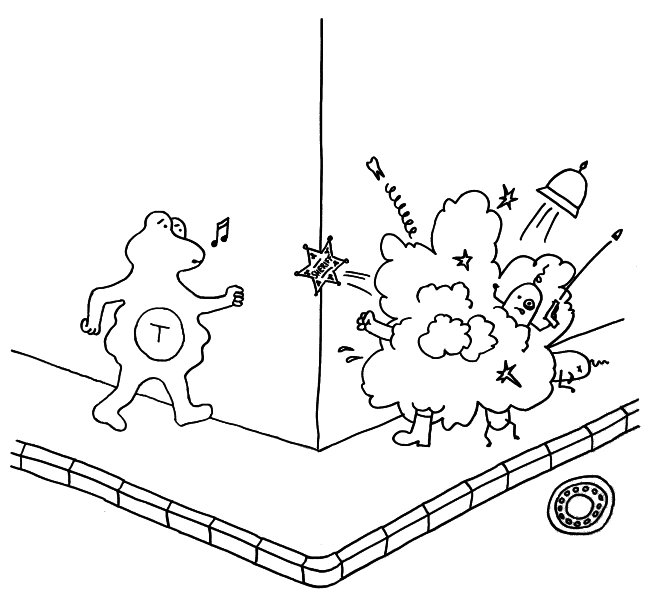
Am Ende eines viertelstündigen Vortrags, der unten verlinkt ist, fasst Matzinger ihre Vorstellung vom Immunsystem in einem eingängigen Bild zusammen: Es besteht ihr zufolge nicht etwa aus schießwütigen Cops, die auf jeden losgehen, den sie nicht spätestens in der Highschool kennen gelernt haben …

… sondern aus Feuerwehrleuten im Bereitschaftsdienst, die in der Einsatzzentrale in aller Ruhe Karten spielen, bis ein Alarm eingeht. Dann rücken sie aus und löschen den Brand. Dabei ist es ihnen völlig egal, ob ein Nachbar die 112 angerufen hat, ein durchreisender Vertreter oder sogar der Brandstifter selbst.

Lange litt Matzingers „danger theory“ darunter, dass sie selbst keine Experimente durchführt, sondern eher Theoretikerin ist. Und DAMPs waren verdammt schwer nachzuweisen. Inzwischen hat man aber einige sehr überzeugende Beispiele gefunden. Eines davon, ein Protein mit dem sperrigen Kürzel HMGB1, werde ich im nächsten Beitrag vorstellen. Es übt zahlreiche Funktionen aus, reguliert seine Aktivität auf faszinierende Art und Weise selbst und spielt bei mehreren Autoimmunerkrankungen eine unglückliche Rolle: Es tritt aus den vom fehlgeleiteten Immunsystem beschädigten Zellen aus und treibt dann den Teufelskreis an.
Linktipps
Polly Matzinger (2001): The Danger Model in Its Historical Context
Polly Matzinger (2002): The Danger Model: A Renewed Sense of Self (PDF)
Video: Polly Matzinger Recaps 65 Years of Immunological Theory in One Diagram (2016, 16 Minuten)