#NaNoWriMo22, Tag 7 (an Tag 6 habe ich Vorarbeiten für diesen Artikel erledigt)
Tiefseeanglerfische und Seenadeln – also die langgestreckten Grasnadeln, die gekrümmten Seepferdchen und ihre Verwandtschaft – haben auf den ersten Blick nicht viel gemeinsam, einmal abgesehen davon, dass sie Fische sind: Die Tiefseeangler (Ceratioidei) leben allesamt in der finsteren Tiefsee und gelten aufgrund ihres ungewöhnlichen Körperbaus mit den riesigen Mäulern, den vorgeschobenen Unterkiefern und den spitzen Zähnen als hässlich, ja monströs – was allerdings auch daran liegt, dass die meisten Exemplare, die wir hier oben zu Gesicht bekommen, durch die Dekompression regelrecht zermatscht sind. Die Seenadeln (Syngnathidae) bevorzugen lichtdurchflutete Seegraswiesen im Flachwasser, haben winzige Mundöffnungen und wirken auf uns grazil und einnehmend.
Auch ihr Liebesleben ist auf den ersten Blick grundverschieden: Seenadelmännchen sind nicht winzig klein und wachsen nicht an einem Weibchen fest. Dafür werden sie trächtig! Das Spektrum reicht von einem einfachen Festkleben der Eier, die ihnen die Partnerin übergibt, am Bauch oder unter dem Schwanz, bis zum Austragen in einer komplett geschlossenen Bruttasche, einem veritablen Schwangerschaftsbauch wie in meinem Cartoon. Der männliche Organismus behütet und nährt den Nachwuchs, der zu diesem Zweck in ein schwamm- oder placentaartiges Gebilde eingebettet wird. Und der Vater stattet die Kleinen auch mit einem immunologischen Starter-Kit aus, sodass sie die vielen Bakterien und anderen potenziellen Krankheitserreger sofort bekämpfen können, wenn sie bei der Geburt aus dem schützenden Bauch ins weite Meer entlassen werden.
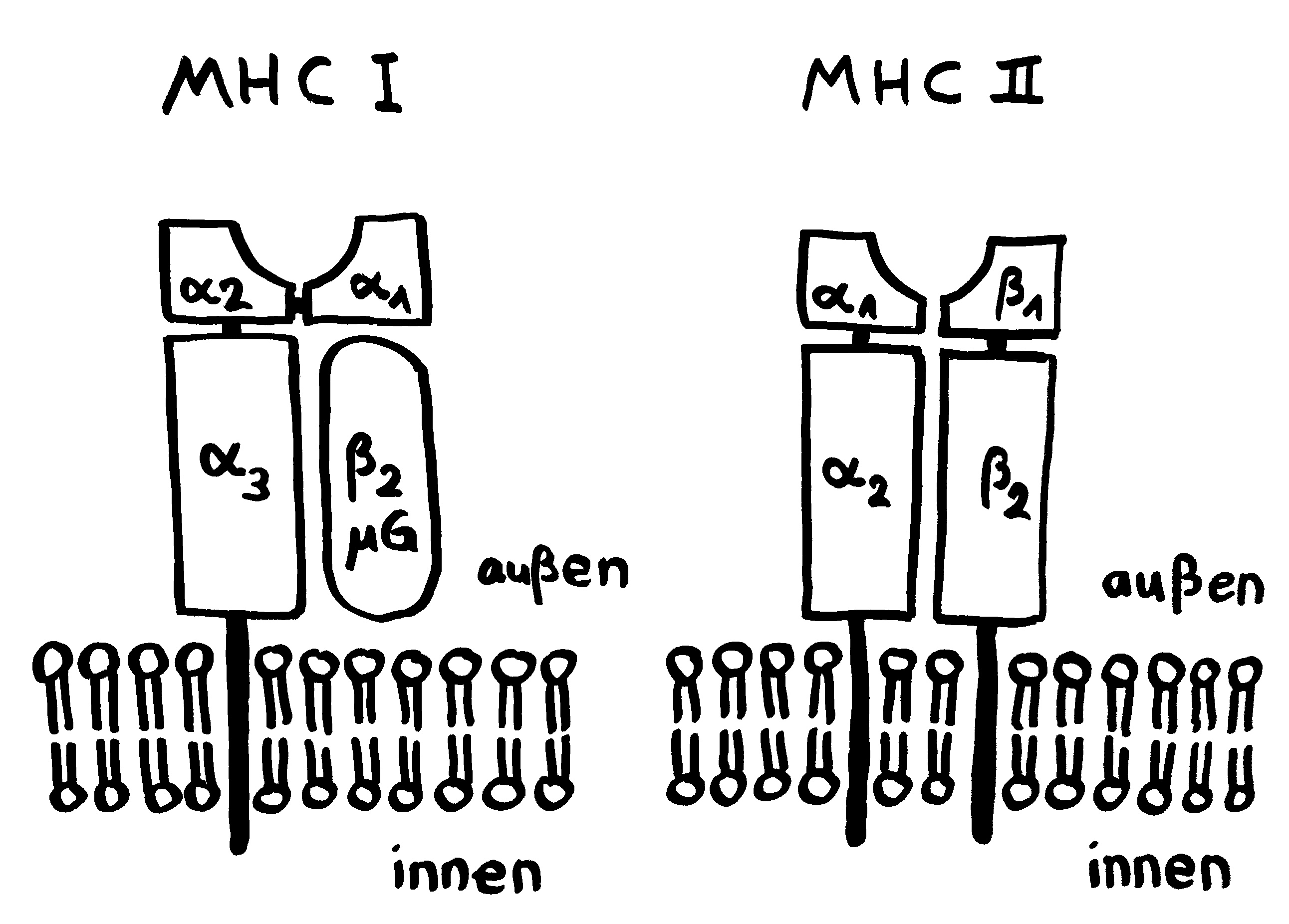
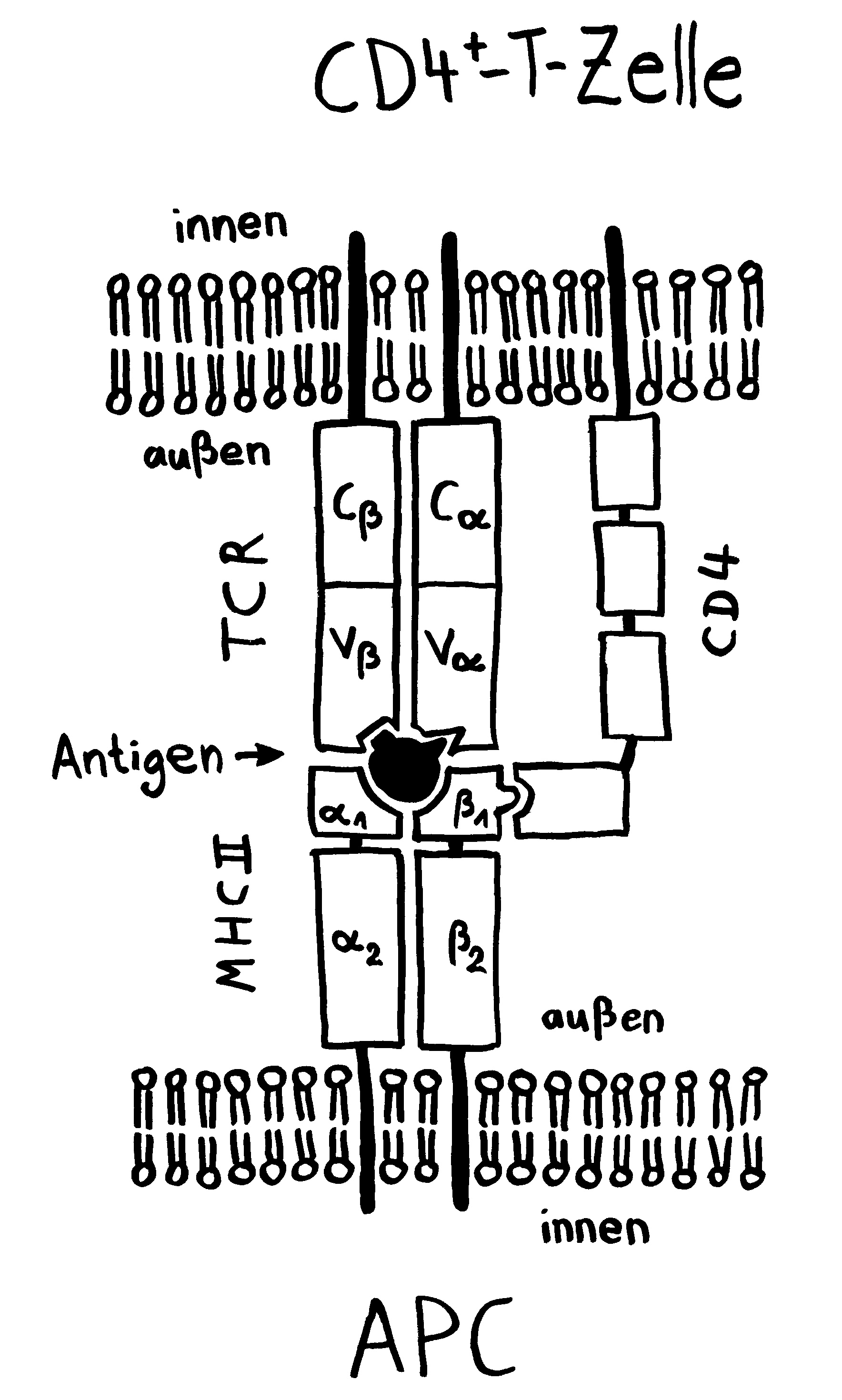
Und da taucht sie dann auf, die Parallele zu den Tiefseeanglern: Wie kommt es, dass das Immunsystem männlicher Seenadeln die Eier bzw. Embryonen nicht abstößt? Sie sind für den väterlichen Organismus doch hemiallograft, bestehen also zur Hälfte aus fremdem Gewebe, nämlich solchem mütterlichem Ursprungs. Normalerweise müssten sie – wie Transplantate – als „Nicht-Selbst“ bekämpft werden. Bei uns Säugetieren geschieht das nur deshalb nicht, weil zwischen der mütterlichen Gebärmutter und dem embryonalen Gewebe eine lückenlose Schutzschicht eingezogen ist, die keine MHC-Komplexe auf der Oberflächen trägt, sodass die mütterlichen Immunzellen gewissermaßen blind sind für das fremde Gewebe, das sich hinter diesem Wall verbirgt. (Das ist jetzt arg verkürzt dargestellt, soll hier aber reichen.)
Ein Forschungsteam um Olivia Roth am GEOMAR Helmholtz Centre for Ocean Research in Kiel untersucht die Schwangerschaften der Seenadeln seit über zehn Jahren, um unter anderem dieses Rätsel zu lösen. Dank des schon erwähnten breiten Spektrums (vom äußerlichen Festkleben der Eier am Bauch über offene Bruttaschen oder -rinnen bis zu geschlossenen Taschen, deren kleine Öffnung das Männchen erst zur Übernahme der Eier und später zur Geburt des ausgetragenen Nachwuchses kontrolliert öffnen kann) lässt sich hier einiges über die Evolution der Trächtigkeit lernen, das dem bloßen Studium der Säugetiere nicht zu entnehmen wäre. Denn bei den Säugetieren laufen alle Schwangerschaften im Grunde ähnlich ab; sie sind gewissermaßen alle vollkommen; „ein bisschen schwanger“ gibt es hier nicht!
Anders bei den Seenadeln. Hier zeigt sich: Je inniger der Kontakt zwischen väterlichem Organismus und Nachwuchs, desto stärker ist die Art „immundefizient“. Wie bei den Tiefseeanglern fehlen den Seepferdchen der Gattung Hippocampus und den Grasnadeln der Gattung Synghathus diejenigen Teile der erworbenen Abwehr, die eine Abstoßungsreaktion auslösen könnten. Die Gene im Haupthistokompatibilitätskomplex der Klasse II (MHC II) sind bei Syngnathus typhle abhanden gekommen oder defekt. Auch T-Zellen des Typs CD4+ scheinen bei dieser Art zu fehlen; sie hat also keine T-Helferzellen.
Andere Komponenten des Immunsystems sind zwar vorhanden und im Prinzip funktionstüchtig, werden aber bei Männchen während der Schwangerschaft herunterreguliert, um ihre immunologische Toleranz gegenüber den Halb-Fremdlingen in der Bruttasche zu erhöhen. Das gilt zum Beispiel für den Haupthistokompatibilitätskomplex der Klasse I (MHC I).
Eine Untersuchung der Entzündungsparameter bei Syngnathus typhle ergab deutliche Parallelen zu Säugetieren wie uns Menschen: Ganz zu Beginn einer Schwangerschaft, bei der Einnistung, tritt bei Grasnadeln wie Säugern eine lokale Entzündung auf, ohne die das zur Einbettung des befruchteten Eies nötige Gewebe gar nicht entstehen kann. Entzündungen fördern ja unter anderem die Bildung von Blutgefäßen. Dann folgt eine längere entzündungsfreie Phase, in der das elterliche Immunsystem maximale Toleranz übt. Schließlich steigen die Entzündungsparameter wieder an, denn die Geburt ist im Grunde nichts anderes als eine verspätete Abstoßung, ja ein Ausstoßen der nunmehr auch außerhalb des elterlichen Körpers lebensfähigen Kinder. Auch viele andere Gene, die bei uns Säugern in der Schwangerschaft je nach Phase stärker oder schwächer abgelesen werden als sonst, durchlaufen bei den Grasnadeln dieselbe Dynamik.
So, und warum interessiert mich das, warum gehört es ins Autoimmunbuch? Die meisten Autoimmunerkrankungen sind bei Frauen häufiger als bei Männern, teilweise extrem viel häufiger. Dafür werden in der Wissenschaft mehrere mögliche Ursachen diskutiert. Es könnte etwa an den weiblichen und männlichen Hormonen liegen, an den unterschiedlichen Genen auf dem X- und dem Y-Chromosom – oder am sogenannten X-Dosis-Effekt, also daran, dass Frauen zwei X-Chromosomen haben, Männer aber nur eines. Hinzu kommen Umweltfaktoren, etwa eine unterschiedliche Ernährung, unterschiedliche Berufe, unterschiedliche Kosmetika usw. Diese möglichen Ursachen schließen einander nicht aus, sondern könnten auch in Kombination miteinander das Erkrankungsrisiko herauf- oder herabsetzen.
Beim Menschen lassen sich diese Faktoren aber oftmals nicht sauber voneinander trennen: Das Geschlecht mit den beiden X-Chromosomen ist zugleich dasjenige, das Eizellen hervorbringt, die relativ groß sind und zum Beispiel Mitochondrien und Moleküle des Immunsystems enthalten. Und wer die Eizellen hervorbringt, trägt auch die Kinder aus – von Leihmutterschaften usw. einmal abgesehen. Aber auch Studien, die relativ einfach und ethisch unbedenklich wären, fehlen ärgerlicherweise. So wird seit Jahrzehnten spekuliert, ob eine Mutterschaft das Risiko für Hashimoto-Thyreoiditis erhöht, weil eine schlummernde Veranlagung während des immunologischen und hormonellen „Ausnahmezustands Schwangerschaft“ zum Ausbruch kommen könnte. Aber gute Statistiken dazu, also etwa Hashimoto-Prävalenzen bei 50-jährigen Frauen mit 0, 1, 2 oder 3 Kindern, habe ich noch nie gesehen!
Die Seenadeln bieten hier eine einmalige Chance: Es gibt ein männliches Geschlecht, das wie üblich zahlreiche winzige Spermien hervorbringt, und ein weibliches, das relativ wenige große, ressourcenreiche Eizellen produziert. Aber danach sind die Rollen vertauscht. Wie unterscheidet sich das Immunsystem der Männchen von dem der Weibchen – generell und insbesondere in den verschiedenen Phasen der männlichen Trächtigkeit? Geschlechtsspezifische Auswertungen habe ich in der Literatur noch nicht entdeckt, aber das kommt sicher noch.
Auch hier im Blog bleiben wir in den nächsten Tagen und Wochen noch beim Thema Fortpflanzung. Denn in der riesigen, bunten Klasse der Knochenfische (und auch bei ihren Cousins, den Knorpelfischen) gibt es offenbar fast nichts, was es nicht gibt. Freuen wir uns also auf die Reisfische, bei denen die Weibchen ihre Jungen im Geschlechtstrakt herumtragen, ohne im engeren Sinne schwanger zu sein. Und auf die Maulbrüter, bei denen teils beide Geschlechter, teils aber nur die Weibchen den Nachwuchs im Maul behüten, was sich wiederum deutlich in ihrem Immunsystem niederschlägt.
Literatur:
GEOMAR (2012): Der Beitrag der Väter. Wie männliche Fische das Immunsystem ihrer Nachkommen aktivieren können. Pressemitteilung.
Roth et al. (2020): Evolution of male pregnancy associated with remodeling of canonical vertebrate immunity in seahorses and pipefishes. Forschungsarbeit, Open Access.
Parker et al. (2021): Immunological tolerance in the evolution of male pregnancy. Forschungsarbeit, Open Access.