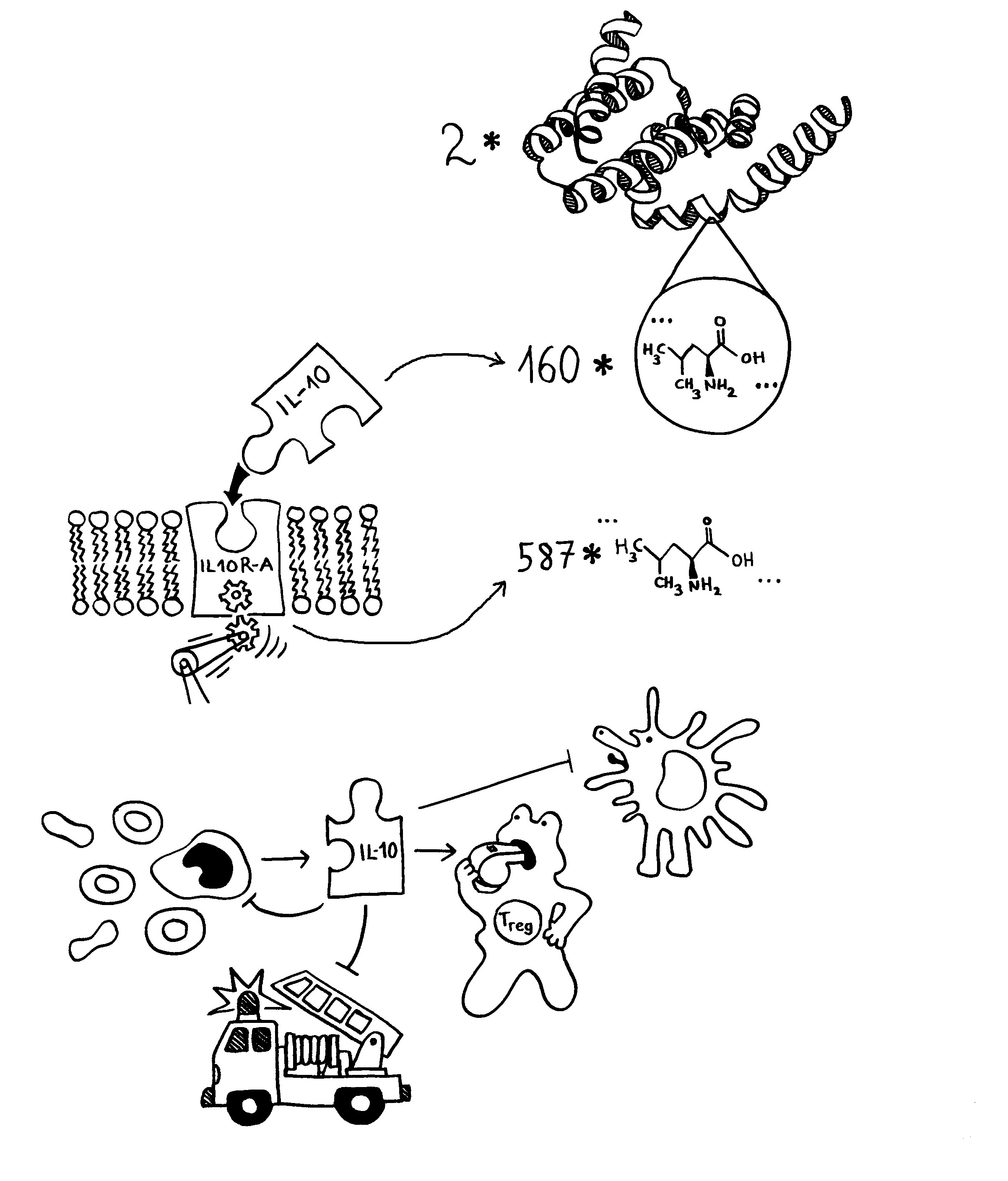
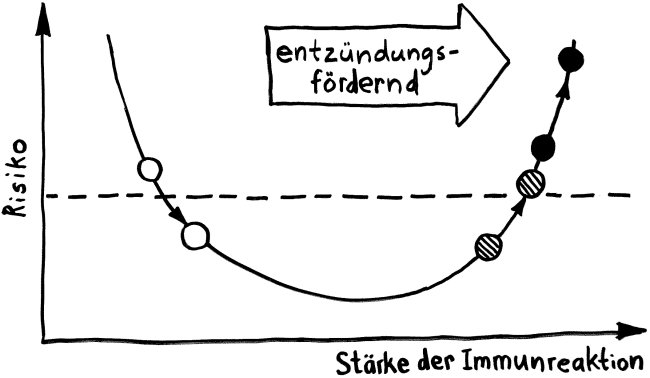
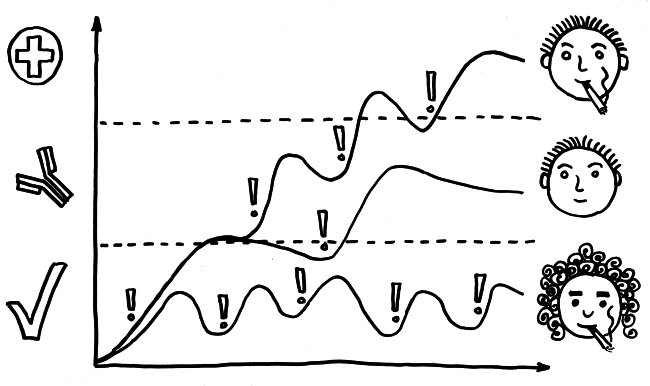
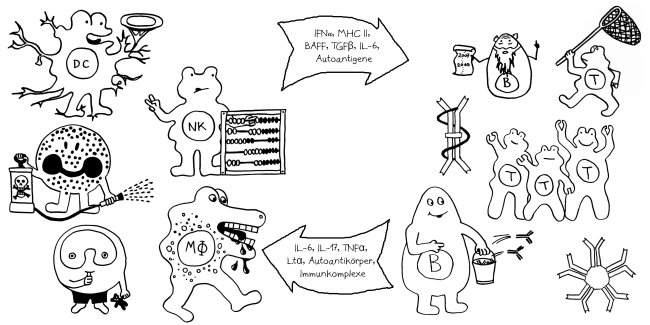
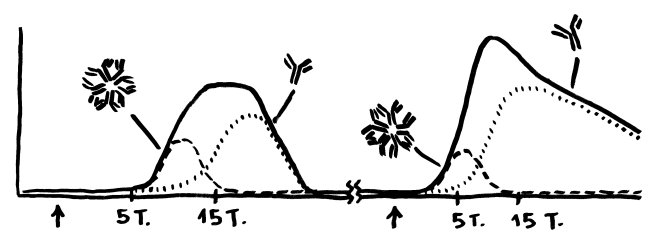
Schon länger ist bekannt, dass die meisten genetischen Varianten, die das Risiko für Autoimmunerkrankungen erhöhen, gar nicht im abgelesenen Bereich irgendeines Gens liegen, sondern in nicht codierenden, rein regulatorischen DNA-Sequenzen, die das Ausmaß beeinflussen, in dem andere Sequenzen abgelesen werden. Durch sogenannte genome-wide association studies (GWAS) kennen wir Aberhunderte solcher Einzelnukleotid-Polymorphismen (single nucleotide polymorphisms oder SNPs), also Stellen in unserem Genom, an denen bei einem Teil der Menschen eine Nukleobase (A, T, C oder G) durch eine andere ersetzt ist.
Diese winzigen Varianten verändern die Feinstruktur der DNA-Doppelhelix und der Nukleosomen, also der kabelrollenartigen Proteinkomplexe, um die die Doppelhelix gewickelt ist. Dadurch können zum Beispiel andere Proteinkomplexe, die für die Ablesung von Genen, also die Produktion von Messenger-RNA-Strängen verantwortlich sind, besser oder schlechter an die DNA andocken und sich von dort aus an ihren Einsatzort weiterhangeln. Oder die DNA bildet Schleifen, durch die eine aktivierende Sequenz mit einer aktivierbaren Sequenz in Kontakt kommt, usw. Die meisten dieser Regulierungsmechanismen sind noch nicht richtig aufgeklärt.
Bis vor kurzem konnte man auch nicht sagen, welches Gen nun durch einen SNP stärker oder schwächer abgelesen wird als sonst. Es kann das nächste oder übernächste Gen in die eine oder in die andere DNA-Strang-Richtung sein (cis-Regulierung) oder irgendein Gen in großer Entfernung, sogar auf einem anderen Chromosom (trans-Regulierung). Das lässt sich neuerdings durch aufwändige Analysen der sogenannten expression quantitative trait loci – kurz: eQTLs – aufklären. Das sind DNA-Sequenzen, die die Ablesung eines Gens nicht einfach ein- oder ausgeschalten, sondern hoch- oder herunterregulieren – und genau das ist bei vielen Genen des Immunsystems der Fall. (Das quantitative Merkmal ist hier also nicht, wie bei klassischen QTLs, ein kontinuierlich variierendes äußerliches Merkmal wie etwa die Körpergröße, sondern die mRNA-Menge, die bei der Transkription des Zielgens entsteht.)
Die eQTL-Analysen unserer Immunsystem-Gene sind in den letzten Monaten immer genauer geworden: Anfangs hat man untersucht, wie sich SNPs, die statistisch mit Autoimmunerkrankungen in Verbindung gebracht wurden, auf die Ablesestärke von Immunsystem-Genen in einem Gemisch aller möglicher weißer Blutkörperchen (Monozyten, T-Zellen, B-Zellen usw.) auswirken. Das ist ungefähr so, als würde man ein Elektroenzephalogramm von einer ganzen Gruppe von Leuten machen und dann aus dem Gewirr von Signalen darauf rückschließen wollen, welche Prozesse in ihren Gehirnen ablaufen (Yao et al. 2014):
 Als Nächstes hat man einzelne Immunzelltypen aus den Blutproben von gesunden Probanden isoliert und dann beispielsweise die Ablesung bestimmter Immunsystem-Gene in den Monozyten von Europäern mit der Ablesung derselben Gene in den Monozyten von Asiaten verglichen (Raj et al. 2014):
Als Nächstes hat man einzelne Immunzelltypen aus den Blutproben von gesunden Probanden isoliert und dann beispielsweise die Ablesung bestimmter Immunsystem-Gene in den Monozyten von Europäern mit der Ablesung derselben Gene in den Monozyten von Asiaten verglichen (Raj et al. 2014):
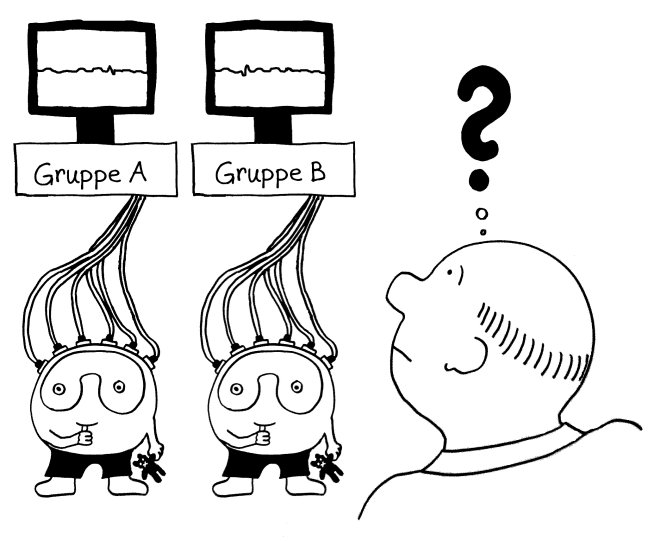
Bei vielen Genen, die man im Verdacht hat, das Risiko für Autoimmunerkrankungen zu beeinflussen, konnte man so allerdings keine großen Unterschiede zwischen den Gruppen entdecken. Das ist kein Wunder, denn die Immunzellen wurden im nicht angeregten Grundzustand untersucht. Immunreaktionen werden aber durch Alarmsignale ausgelöst, zum Beispiel durch die Konfrontation mit Krankheitserregern oder mit Molekülen, die für diese Pathogene typisch sind – etwa Lipopolysaccharide aus Bakterien-Zellwänden.
Also hat man im nächsten Schritt im Rahmen des ImmVar-Projekts bestimmte Zelltypen aus dem Blut unterschiedlicher Probandengruppen isoliert und sie dann mit solchen Gefahrensignalen konfrontiert, um sie zu aktivieren und anschließend ihre Reaktionen zu erfassen (Fairfax et al. 2014, Lee et al. 2014, Ye et al. 2014):
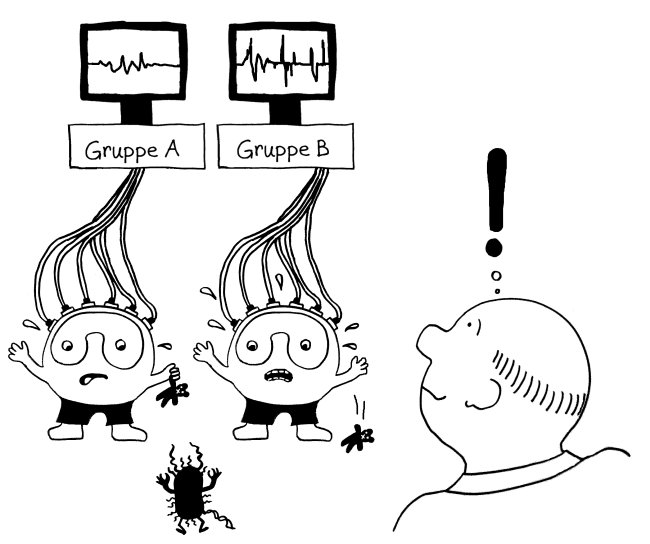
Und siehe da: Bestimmte Gene werden nach der Aktivierung des entsprechenden Immunzelltyps (hier Monozyten) besonders stark abgelesen, wenn die DNA der Probanden an anderer Stelle einen bestimmten Risiko-Genort enthält.
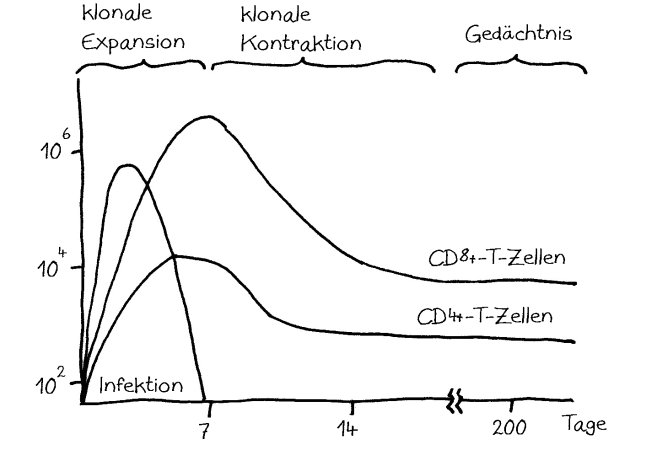
Dabei gab es bereits einige Überraschungen. Beispielsweise wird die Ablesung von Genen, die die Zellvermehrung von Gedächtnis-T-Zellen steuern, durch die bekannten Risiko-Genorte für wichtige Autoimmunerkrankungen wie Typ-1-Diabetes oder rheumatoide Arthritis kaum gesteigert (Hu et al. 2014). Dabei dachte man bisher, diesem Zelltyp käme bei Autoimmunerkrankungen durch seine übermäßige Vermehrung und Aktivierung eine Schlüsselrolle zu.