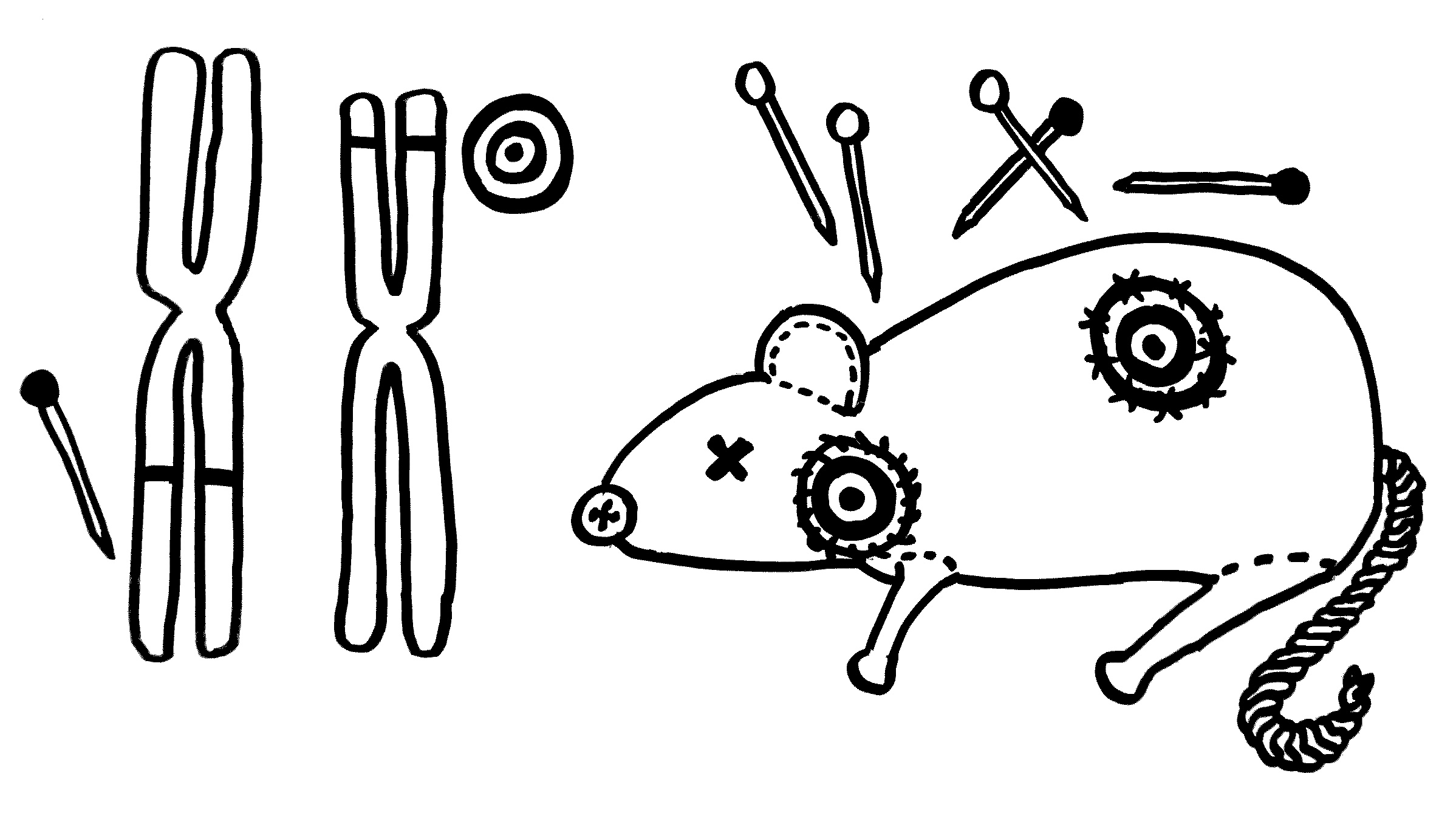
Zwei neue Skizzen fürs Buch, inspiriert durch An Goris und Adrian Liston, „The immunogenetic architecture of autoimmune disease„, 2012 (Open Access):
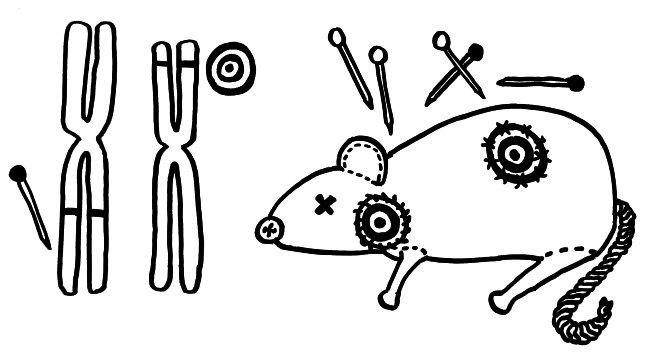
Nur wenige Autoimmunerkrankungen folgen einem einfachen Mendel’schen Erbgang. Meist sind zahlreiche Genvarianten beteiligt, die das Erkrankungsrisiko für sich genommen – wenn überhaupt – nur minimal steigern und erst gemeinsam zum Ausbruch führen. Dabei tragen einige Genvarianten zur allgemeinen Neigung des Immunsystems zu Überreaktionen bei (Voodoo-Nadeln), und andere legen fest, welches Organ betroffen sein wird (Zielscheiben).
NOD-Mäuse wurden als Typ-1-Diabetes-Modell gezüchtet; normalerweise wird ihre Bauchspeicheldrüse durch Autoimmunreaktionen zerstört (Zielscheibe auf dem Rumpf). Wenn man ihr Diabetes-Risikoallel H2g7, das zum HLA-Komplex gehört, durch die Genvariante H2h4 ersetzt, bleiben die Tiere nicht etwa gesund: Sie bekommen eine Schilddrüsen-Autoimmunerkrankung (Zielscheibe am Hals). Auch beim Menschen scheinen die meisten HLA- oder MHC-Klasse-II-Varianten auf dem 6. Chromosom festzulegen, welche Autoantigene und damit welche Organe angegriffen werden, während Risikogenorte an anderen Stellen im Genom darüber entscheiden, ob das Immunsystem überhaupt zu Autoimmunstörungen neigt.
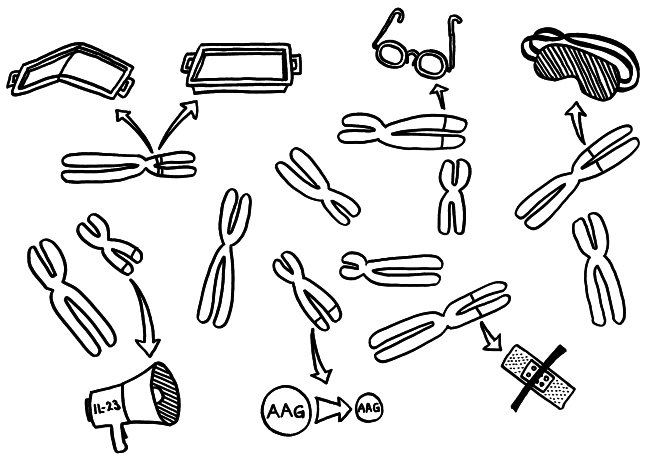
Die Genetik der Autoimmunerkrankungen ist ein etwas undankbares Forschungsfeld, auf dem man nicht hoffen darf, die eine Genvariante zu entdecken, die für einen Großteil der Erkrankungen verantwortlich ist, und daraus eine simple Therapie abzuleiten. Stattdessen kann es sein, dass jemand chronisch krank wird, weil
- eine MHC-Klasse-II-Variante auf Chromosom 6 zu einer schlechten Präsentation eines Autoantigens im Thymus führt, sodass das Immunsystem diesem Autoantigen später nicht gänzlich tolerant gegenüberstehen wird (geknicktes Tablett),
- ein anderes MHC-Klasse-II-Molekül, das auf demselben Chromosom codiert ist, ein Autoantigen besonders stabil bindet, sodass dieses Autoantigen den T-Zellen im Lymphgewebe besonders häufig und lange präsentiert wird, womit die Gefahr einer T-Zell-Aktivierung steigt (tiefes Tablett),
- eine seiner Genvarianten zu besonders scharfsichtigen T-Zell-Rezeptoren führt, sodass die T-Zellen bei einer Präsentation des passenden Autoantigens besonders leicht aktiviert werden (Brille),
- eine andere Genvariante die regulatorischen T-Zellen (Tregs), die überzogene Immunreaktionen normalerweise ausbremsen, träge oder blind macht (Schlafmaske),
- ein weiteres Risikoallel in aktivierten Immunzellen zu einer ungewöhnlich starken Produktion entzündungsfördernder Zytokine führt, die dann immer weitere Immunzellen anlocken (Megafon),
- wieder ein anderes Risikoallel die Expression bestimmter Autoantigene im Thymus schwächt, sodass das Immunsystem ihnen gegenüber nicht tolerant gestimmt wird (geschrumpftes AAG) und
- eine Genvariante an noch einem anderen Genort die Wundheilung in einem Organ hemmt, das durch einen Autoimmunprozess beschädigt wurde (Pflaster).
Auch diese Darstellung der Polygenie der Autoimmunerkrankungen ist noch stark vereinfacht – von den Wechselwirkungen zwischen unseren Genprodukten und dem Mikrobiom, unserer Nahrung, Krankheitserregern und weiteren Umweltfaktoren einmal ganz abgesehen.
Wenn also der nächste Wunderheiler um die Ecke kommt, der behauptet, man müsse nur ein bestimmtes Vitamin weglassen oder ein Mineralpräparat zu sich nehmen, um von einer nahezu beliebigen Autoimmunerkrankung geheilt zu werden: bitte auslachen.