
„Nichts in der Biologie ist sinnvoll außer im Lichte der Evolution“, schrieb der Evolutionsbiologe Theodosius Dobzhansky 1973: Ohne dieses Licht bleibe die Biologie ein Haufen unzusammenhängender Fakten, die kein stimmiges Bild ergeben.
Es lohnt sich, Widersprüchen zwischen Tatsachen und etablierten Vorstellungen nachzuspüren und dabei die Evolution als Richtschnur zu nehmen – also das Wechselspiel von Mutation und Selektion, durch das sich Arten in ihrer Umwelt, aber auch bestimmte Zellpopulationen in unserem Körper entwickeln.
Merkwürdigkeiten
Ein solcher Widerspruch ist Peto’s paradox: 1975 wies der Epidemiologe Richard Peto darauf hin, dass große Säugetiere wie Blauwale oder Elefanten trotz ihrer erheblich höheren Zellzahl und ihrer Langlebigkeit nicht wesentlich früher oder häufiger an Krebs erkranken als kleine, kurzlebige Arten wie Mäuse.
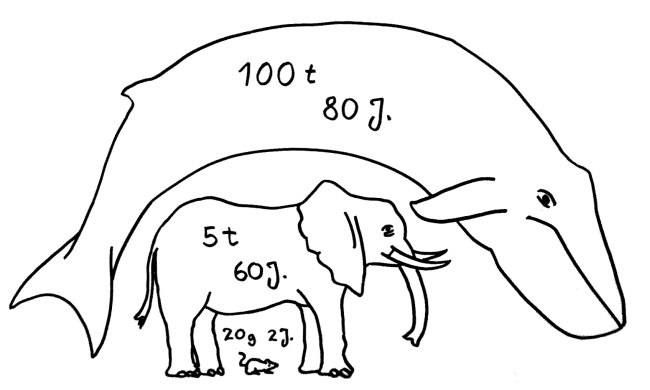
Petos Paradox: Obwohl Wale und Elefanten mehr Zellen haben und älter werden als Mäuse, sterben sie nicht wesentlich öfter an Krebs.
Das passt nicht recht zu dem allgemein akzeptierten Mehrstufenmodell der Krebsentstehung. Nach diesem müsste Krebs nämlich ausbrechen, sobald sich in einer Zell-Linie nacheinander mehrere zufällige Mutationen ereignet haben, die zusammen zu einer unkontrollierten Zellvermehrung führen.
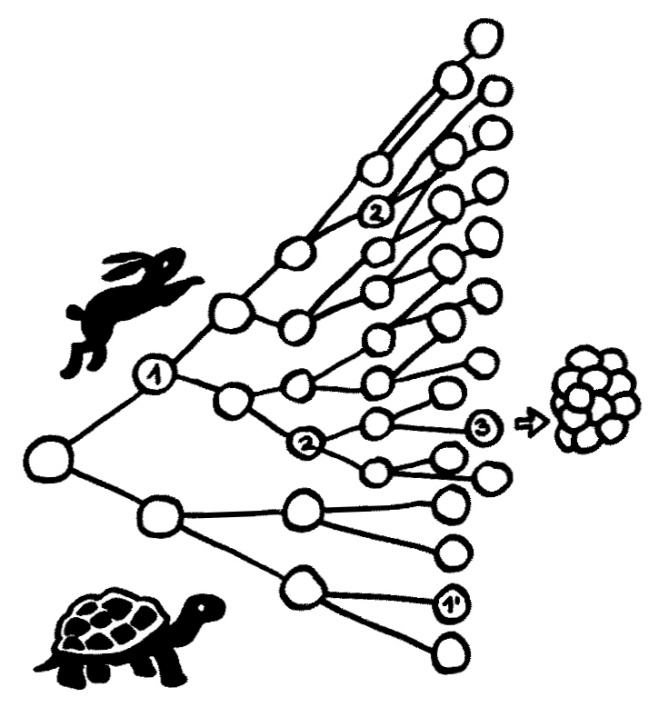
Wenn Krebs ausbricht, sobald bestimmte Mutationen zusammenkommen, müssten Zellen, die sich schneller teilen (oberer Zweig), krebsanfälliger sein. Aber das ist nicht immer der Fall.
Eine zweite Merkwürdigkeit ist der oftmals späte Ausbruch von Krebs beim Menschen, typischerweise nach dem Ende der Reproduktionsphase, und zwar über die meisten Krebsarten und betroffenen Organe hinweg – ob es dort nun viele oder wenige Stammzellen gibt, aus denen die ersten Krebszellen hervorgehen, und ob sich diese Stammzellen nun häufig oder selten teilen. Denn nach dem Mehrstufenmodell müsste Krebs in Organen mit großem Stammzellpool und hohen Zellteilungsraten früher ausbrechen, da sich die tumorbildenden Mutationen dort früher anhäufen sollten – genau wie in größeren Tieren.
Was ist Krebs überhaupt?
Zum besseren Verständnis des Mehrstufenmodells und seiner Unzulänglichkeit blenden wir die beträchtlichen Unterschiede zwischen all den Krebsformen und individuellen Verläufen einmal aus und betrachten das große Ganze: Was ist Krebs?
Wenn im Erbgut einer Zelle etwas schief gelaufen, etwa bei der letzten Zellteilung ein Kopierfehler aufgetreten ist, gibt es mehrere Möglichkeiten. Die DNA kann von Enzymkomplexen im Zellkern repariert werden. Oder die Zelle kann unschädlich gemacht werden – entweder durch einen Ruhestandsmodus namens Seneszenz, in dem sie sich insbesondere nicht weiter teilt, oder durch ein Selbstmordprogramm namens Apoptose, das auch die Beseitigung der Überreste durch Zellen des Immunsystems einschließt.
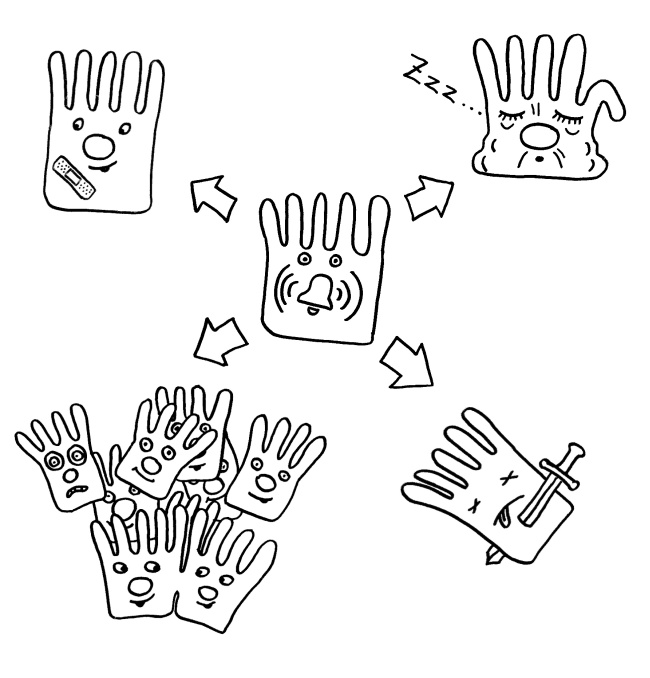
Eine Zelle, in deren Erbgut etwas schief gelaufen ist (Alarm, Mitte), kann entweder repariert werden oder in Seneszenz verfallen oder Selbstmord begehen oder – wenn diese Schutzmechanismen versagen – zur Krebszelle werden.
Versagen diese Mechanismen, teilt sich die defekte Zelle unter Umständen unkontrolliert weiter. So entstehen im Gewebe Tumoren, also bösartige Geschwulste, oder im Fall von Blutzellen Blutkrebs. (Im Folgenden geht es überwiegend um Tumoren.)
Fatale genetische Fehler treten verstärkt in alten Zell-Linien auf. Das liegt an der Verkürzung der Telomere, der Schutzkappen an den Enden der Chromosomen, bei jeder Zellteilung. Telomere bestehen aus den Enden der DNA-Doppelhelix, in denen keine Gene oder Steuersequenzen codiert sind, sondern „Blindtext“ steht, und einigen Proteinen. Dieser Blindtext ist nötig, weil die Enzyme, die vor einer Zellteilung die Chromosomen verdoppeln, beim Ablesen und Nachbauen der DNA-Sequenz auf der Doppelhelix wie auf einer Eisenbahnschiene vorankriechen und sich dabei gewissermaßen selbst im Weg stehen. Sie können das allerletzte Stück der Sequenz nicht nachbauen und hören einfach etwas früher auf.
Aber das geht nur so lange gut, bis eine kritische Minimallänge der Telomere erreicht ist. Danach werden weitere Zellteilungen unterbunden, bevor die Kopiermaschine wichtige genetische Informationen beschneidet. Die Zellen tun noch eine Weile ihre Arbeit und sterben dann ab. Es sei denn, es handelt sich um Stammzellen: Diese zeichnen sich durch eine nahezu unendliche Teilungsfähigkeit aus. Ihre Telomere erreichen nicht die kritische Mindestlänge, da ein Enzym namens Telomerase die verloren gegangenen Basensequenzen durch neuen Blindtext ersetzt.

Die Telomere an den Enden der Chromosomen enthalten keine Erbinformation, sondern Blindtext. Sie werden bei jedem DNA-Kopiervorgang, also jeder Zellteilung kürzer, da der Kopierer selbst Raum einnimmt. Sobald sie zu kurz werden, gibt es einen Zellteilungsstopp – es sei denn, das Enzym Telomerase verlängert die Telomere wieder.
Das ist eine riskante Strategie, denn so können im schlimmsten Fall auch auch genetische Fehler unendlich multipliziert werden, die dem Organismus schaden. Daher halten sich Stammzellen in besonders gut geschützten Nischen auf, etwa im Knochenmark oder in den Krypten (Taschen in der Darmschleimhaut): Hier ist das Risiko von Mutationen geringer als an einer Oberfläche, die sogenannten Mutagenen, also Mutationen auslösenden Einflüssen ausgesetzt sind – etwa toxischen Substanzen, Krankheitserregern oder ultravioletter Strahlung. Außerdem wird die Vermehrung von Stammzellen durch eine Vielzahl von Wachstums- und Hemmstoffen strikt reguliert, in ständiger Rückbindung an den Bedarf an entsprechenden neuen Zellen.
Douglas Hanahan und Robert A. Weinberg haben acht Kennzeichen von Krebszellen sowie zwei krebsbegünstigende Bedingungen zusammengestellt. Demnach sind Krebszellen im Unterschied zu anderen Zellen
- nicht auf externe Wachstumsfaktoren angewiesen,
- unempfindlich für Wachstumsstoppsignale,
- nicht für Selbstmord-Befehle empfänglich,
- unbegrenzt teilungsfähig und damit potenziell unsterblich;
- ihr Stoffwechsel ist verändert, und sie können
- sich der Zerstörung durch das Immunsystem entziehen,
- die Bildung von Blutgefäßen anregen sowie
- aus ihrer Umgebung auswandern und in einem neuen Umfeld Metastasen bilden.
Die beiden krebsfördernden Faktoren sind
- genetische Instabilität und damit eine erhöhte Mutationsrate sowie
- chronische Entzündungen.
Mit einer Verlängerung der Telomere und der dadurch erreichten unbegrenzten Teilungsfähigkeit der Zellen ist es also bei weitem nicht getan: Die Veränderungen, die der Entstehung, Versorgung und Verbreitung von Tumoren im Körper vorangehen oder sie begleiten, sind nur durch eine Vielzahl von Mutationen zu erreichen.
Tumoren: komplexe Produkte der somatischen Evolution
Ein Tumor enthält typischerweise eine Milliarde (10 hoch 9) bis eine Billion (10 hoch 12) Zellen, die im Lauf von einigen 100 oder 1000 Zellteilungszyklen entstanden sind. Er besteht aus mindestens zehn, zumeist aber Hunderten oder Tausenden von Klonen: den Nachkommen von ebenso vielen Zellen, in denen sich unterschiedliche Mutationen ereignet haben. Diese Klone sind typischerweise sehr ungleich groß, weil einige wenige Mutationen (die sogenannten Treibermutationen) zumindest zeitweilig einen Selektionsvorteil mit sich bringen, während die übrigen Mutationen entweder selektionsneutral oder nachteilig sind.
Würde man denselben Tumor über längere Zeit mehrmals untersuchen, so würde man immer wieder andere klonale Zusammensetzungen ermitteln, da sich die Selektionsbedingungen mit der Zeit ändern. So kann eine Mutation in einem jungen, kleinen Tumor nachteilig oder neutral sein, aber ihrem Zellklon später im Inneren eines größeren Tumors einen Überlebensvorteil verschaffen, weil dort Sauerstoffmangel herrscht. Die Zellen in einem Tumor konkurrieren ja miteinander um Ressourcen: um Nährstoffe, Sauerstoff, Raum, Wachstumsfaktoren und Überlebenssignale, die alle zusammen ihre ökologische Nische bilden.
Dieser dynamische Mutations- und Selektionsprozess wird als somatische (also in einem Körper ablaufende) Evolution bezeichnet. Wir kennen ihn aus einem anderen Zusammenhang: aus der Entwicklung einer schlagkräftigen erworbenen Immunabwehr, insbesondere der Affinitätsreifung der B-Zellen. Somatische Evolution ist also nicht per se pathologisch, genau wie die oben aufgelisteten Merkmale von Krebszellen in anderen Zusammenhängen unauffällig, ja lebensnotwendig sein können – etwa bei der Wundheilung.
Der Evolutionsmechanismus ist derselbe wie bei der Darwin’schen Evolution der Arten. Dass die Tumoren ihren Träger oder Wirt letzten Endes meist umbringen, also sozusagen langfristig gegen die eigenen Interessen handeln, steht dem nicht entgegen: Auch die Evolution der Arten läuft nicht zielgerichtet ab. In Grunde verhält der Mensch sich ähnlich wie Krebs: Mit seinem evolutionären Erfolg beraubt er sich der eigenen Existenzgrundlage, indem er seine Nische zerstört. (Übrigens gibt es ein paar Krebsarten, die nicht gemeinsam mit ihren Wirten zugrunde gehen. Am bekanntesten sind die Tumoren im Gesicht des Beutelteufels, die bei Kämpfen mit Artgenossen wie eine Infektion auf einen neuen Wirt überspringen und den Bestand der Art massiv gefährden. Ein ähnliches Phänomen gibt es bei Hunden. Und kürzlich entdeckten Forscher ansteckenden Krebs bei Muscheln, der sogar die Artgrenze übersprungen hat, heute also in anderen Muscheln verbreitet ist als jenen, in denen er entstand.)
Mutationen: Umbau in großem Stil
Unter „Mutation“ stellt man sich oft winzige Veränderungen im Erbgut vor, klassischerweise sogenannte Punktmutationen: die Hinzufügung, den Wegfall oder den Austausch einzelner Basenpaare. Es ist aber gar nicht klar, ob solche Punktmutationen in Tumoren überhaupt häufiger anzutreffen sind als in gesundem Gewebe. Weit verbreitet sind dagegen sogenannte Mitose-Katastrophen, also großräumige genetische Instabilitäten: Duplikationen (Verdopplungen), Deletionen (Beseitigungen) und Translokationen (Verlagerungen) ganzer Chromosomen-Abschnitte oder gar ganzer Chromosomen. Wenn mit den Telomeren etwas nicht stimmt, können zum Beispiel die Enden beider Arme eines X-förmigen Chromosoms verschmelzen. Wenn danach einer der Arme in der Nähe des Centromers (des Mittelstücks des X) bricht, entstehen bei der nächsten Zellteilung ein sehr viel größeres und ein sehr viel kleineres Chromosom. Die meisten dieser Zellen sind nicht lebensfähig, aber wenn doch, besteht die Gefahr, dass sie mindestens eines der oben genannten Krebszellen-Merkmale ausprägen.
Hinzu kommen epigenetische Veränderungen, die schätzungsweise um mehrere Größenordnungen häufiger auftreten als genetische Instabilitäten. Sie können innerhalb ihres Tumor-Zellklons erblich sein und beeinflussen den Phänotyp, also das Verhalten von Zellen ebenso wirksam wie Änderungen in der DNA-Basensequenz, indem sie die Ablesung von Genen ermöglichen, verstärken, hemmen oder verhindern.
Das Mehrstufenmodell der Karzinogenese ist zu einfach
Zwar gibt es sogenannte pleiotrope Gene, also solche, die mehrere scheinbar unzusammenhängende Auswirkungen haben. Aber damit eine Zelle zur Krebszelle wird, müssen so viele Merkmale zusammenkommen und so viele Schutzmechanismen überwunden werden, dass man seit den 1950er-Jahren davon ausgeht, dass erst eine ganze Reihe von Mutationen gemeinsam zur Tumor- und Metastasenbildung führen. Das passt auch zu der relativ geringen Zahl von Krebserkrankungen in den ersten Lebensjahrzehnten.
Allerdings hat man sich die Sache lange zu einfach vorgestellt – vor allem, da die Methoden, mit denen Tumorproben analysiert wurden, die räumliche Komplexität und die zeitliche Veränderlichkeit dieser Gebilde verschleiert haben. Platt gesagt: Wenn man zahlreiche Tumorzellen in einen Mixer wirft und die DNA dieses Shakes untersucht, erkennt man nicht, dass sie aus verschiedenen Klonen stammen und wie sich diese Klone zueinander verhalten. So entstand die irrige Vorstellung von einem linearen Ablauf:
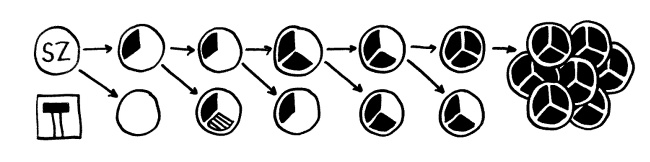
Nur in dauerhaft teilungsfähigen Zellen wie Stammzellen (oben) können sich Mutationen (schwarz) akkumulieren, die schließlich zu Krebs führen. Ausdifferenzierte Zellen, die sich kaum noch teilen, sind Sackgassen (unten).
Richtig ist daran, dass sich die nötigen Treibermutationen nur in dauerhaft teilungsfähigen Zellen ansammeln können, also in echten Stammzellen oder in Krebsvorläuferzellen mit Stammzell-Eigenschaften, etwa verlängerten Telomeren aufgrund einer frühen Treibermutation. Bereits ausdifferenzierte Zellen, die keine oder nur noch wenige Teilungen durchlaufen, sind Sackgassen der Krebs-Evolution.
Dieses Modell hat zwei Schwächen. Ignoriert wurden erstens die Verzweigungen: die Aufspaltung der Klone in Subklone. Man ging davon aus, dass sich eine Treibermutation nach ihrem Auftreten rasch im gesamten Tumor durchsetzt, also ein sogenannter selective sweep die gesamte Zellpopulation durchläuft. Heute weiß man, dass die Zeit zwischen zwei Mutationen meist nicht ausreicht, um die vorige Mutation flächendeckend durchzusetzen – selbst wenn sie einen deutlichen Selektionsvorteil mit sich bringt. Stattdessen sind Tumoren immer komplexer werdende Mosaike aus größeren und kleineren Klonen, in denen neutrale oder leicht nachteilige Mutationen lange mitgeschleppt werden.
Zweitens wurde die Rolle des veränderlichen Selektionsdrucks gegenüber der Bedeutung der Mutationen lange unterschätzt. Denn die Treibermutationen müssen, um sich durchzusetzen, den betroffenen Krebsvorläuferzellen einen Fitnessvorteil verschaffen, sodass sie sich schneller vermehren und damit größere Klone bilden als die anderen Zellen in der Nachbarschaft. In den größeren Klonen ereignen sich dann mit höherer Wahrscheinlichkeit die nächsten Treibermutationen als in den kleinen. Aber auch ein kleiner Klon, der sich lange Zeit nur mit Ach und Krach am Leben hält, kann in der nächsten Entwicklungsstufe des Tumors plötzlich die Oberhand gewinnen – wie hier der mit X gekennzeichnete Klon, der auf die Mutation M3 zurückgeht und sich anfangs kaum vermehren kann, aber durch das Selektionsereignis S2 zum Zuge kommt:
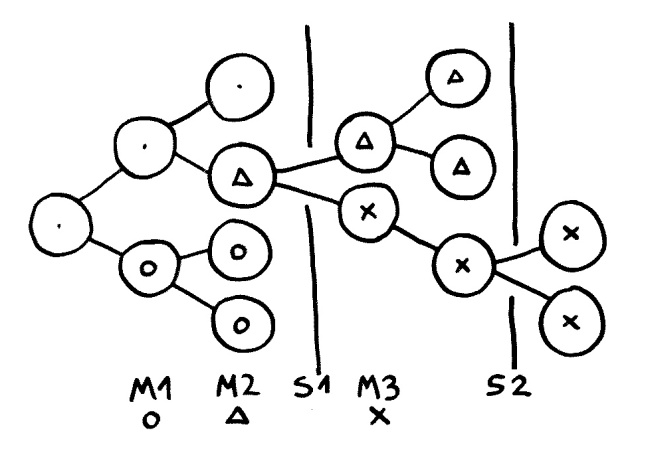
Welche Zellklone überleben und sich ausbreiten, hängt vom aktuellen Selektionsdruck (S1, S2) ab. So kann am Ende ein Klon das Rennen machen, der lange keinen Selektionsvorteil hatte und sich daher kaum vermehrt hat (Mutation M3).
So sind etwa genetische Veränderungen, die eine Zelle zum Kriechen befähigen, in einem primären Tumor unnütz und wahrscheinlich sogar nachteilig, da so Ressourcen an eine nicht genutzte Fähigkeit verschwendet werden, während die Konkurrenz mit denselben Ressourcen schneller wächst und sich teilt. Aber sobald die Lebensbedingungen im primären Tumor unerträglich werden, haben diese Zellen plötzlich einen Selektionsvorteil, denn sie können ihr Bündel packen und über die Blut- und Lymphgefäße in andere Organe mit günstigeren Bedingungen auswandern, um dort Absiedlungen zu bilden.
Das erinnert an einen bekannten Modellorganismus der Biologen, den Schleimpilz Dictyostelium discoideum: Normalerweise lebt er einzellig, wie eine Amöbe. Aber wenn die Lebensbedingungen zu harsch werden, kommt ihm seine Fähigkeit zugute, sich zusammenzurotten und Fruchtkörper zu bilden, deren Sporen vom Winde verweht werden und andernorts ihr Glück versuchen.
Wenn wir dem veränderlichen Selektionsdruck und der sich mit ihm wandelnden Fitnesslandschaft im Tumor mehr Beachtung schenken, ist der stark verzögerte Ausbruch von Krebserkrankungen, oft erst Jahre oder Jahrzehnte nach den entscheidenden Mutationen, nicht mehr so rätselhaft.
Karzinogene sind nicht nur Mutagene
Ausdrücke wie Nische, Konkurrenz, Ressourcen oder Fitnesslandschaft zeigen: Das ökologische Denken hält Einzug in die Onkologie. Sogar eine Entsprechung zu Räuber-Beute-Beziehungen gibt es, denn das Immunsystem bekämpft Krebsvorstufen und übt damit einen Predatorendruck auf sie aus: Krebs- und Krebsvorläuferzellen, die sich vor dem Immunsystem verbergen können, haben einen Vorteil gegenüber der Konkurrenz, die für die Immunzellen sichtbar ist und vernichtet wird. Auch Krebstherapien wie Bestrahlung oder Chemotherapien üben einen Selektionsdruck aus, der Darwins künstlicher Zuchtwahl entspricht, und fördern die Entstehung und Ausbreitung therapieresistenter Klone.
Allerdings hinken die üblichen Verfahren zur Einstufung der Karzinogenität von Substanzen, etwa der Ames-Test, hinter dieser Erkenntnis her: Sie prüfen, ob eine Substanz die Mutationsrate erhöht, ignorieren aber ihren Einfluss auf die Umgebung von Krebs- und Krebsvorläuferzellen und damit auf den lokalen Selektionsdruck. So ist Tabakrauch nicht nur DNA-schädigend und damit mutagen; er schwächt auch das Lungengewebe, sodass Krebsvorläuferzellen Platz zur Ausbreitung haben. Damit steigt auch die Wahrscheinlichkeit für Folgemutationen in ihren Klonen und damit für Lungenkrebs.
Ein zweites Beispiel: Im Magen löst der Keim Helicobacter pylori Entzündungen aus, die zu Magenkrebs beitragen – und zwar nicht nur durch die Schädigung des Erbguts von Magenschleimhaut-Stammzellen, sondern auch durch eine Schwächung der Magenschleimhaut, die Krebsvorläuferzellen dann nicht mehr so gut in Schach halten kann.
Um eine Metapher zu bemühen: Der Meteor, der vor 65 Millionen Jahren auf der Erde einschlug, machte den meisten der bis dahin dominierenden Dinosauriern den Garaus, sodass die kleinen Säugetiere, die es bereits gab, sich anschließend durchsetzen konnten. Sie waren an die neuen Umweltverhältnisse besser angepasst als die Echsen. Ganz ähnlich verhält es sich mit Krebsvorläufern und gesundem Gewebe, wenn sich der Selektionsdruck ändert: Modellrechnungen zur Entwicklung von Stammzellpools haben gezeigt, dass eine Veränderung der Umgebung das Wachstum eines Krebs- oder Krebsvorläufer-Klons stärker beschleunigen kann als eine Mutation.
Körpereigene Krebsbekämpfung
Wie stark der Selektionsdruck ist, wir deutlich, wenn man bedenkt, dass sich Krebszellen alle ein bis zwei Tage verdoppeln, Tumoren aber nur alle sechs bis 200 Tage. Die allermeisten Tumorzellen sterben also oder stellen zumindest die Zellteilung ein – sogar in weit fortgeschrittenen, aggressiven Tumoren. Diese körpereigene Krebsvorbeugung und -bekämpfung schafft es bei den allermeisten Menschen, den Ausbruch der Erkrankung auf die postreproduktive Lebensphase zu verschieben, in der sie den Fortpflanzungserfolg nicht mehr schmälern kann.
Die Krebsabwehr ist zum Teil in den potenziellen Krebsvorläufzellen selbst und zum Teil im Organismus oder im umgebenden Gewebe angesiedelt. Die intrinsischen Mechanismen haben wir oben bereits kennen gelernt: Bei Alarmsignalen, etwa nach einer Erbgutschädigung, wird der Schaden repariert, oder die Zelle geht in den Ruhestand oder begeht Selbstmord. Und wenn nach einer gewissen Zahl von Zellteilungen die kritische Länge der Telomere erreicht ist, ist ebenfalls Schluss mit dem Teilen.
Krebs- und Krebsvorläuferzellen verraten sich durch bestimmte Oberflächenmarker, die im Zuge ihrer Entgleisung exprimiert werden. Immunzellen erkennen viele dieser veränderten Zellen und eliminieren sie; auf diesen Mechanismus komme ich im nächsten Artikel zurück. Neben dieser systemischen Abwehr stemmt sich auch die unmittelbare Nachbarschaft, das örtliche Gewebe, der übermäßigen Ausbreitung von Zellklonen entgegen: Es konkurriert mit den entstehenden Tumoren um Sauerstoff, Nährstoffe und Raum. Oftmals hemmen feste Grenzschichten wie Basalmembranen oder Bindegewebskapseln die Ausdehnung. Wie erfolgreich diese Maßnahmen sind, zeigen die zahlreichen harmlosen, weil örtlich begrenzten Wucherungen, die man im Gewebe älterer Menschen findet.
Blühende und verblühte Landschaften
Sowohl die intrinsische als auch die systemische und die nachbarschaftliche Krebsbekämpfung funktioniert am besten im jungen, gesunden Organismus. In ihm haben unmutierte Zellen gegenüber nahezu allen erdenklichen Mutationen einen Selektionsvorteil, denn ihre genetische Ausstattung ist in Jahrmillionen auf die Existenz in einer solchen Umgebung hin optimiert worden. Zu ihr gehören etwa systemische Regulatoren wie Hormone und Wachstumsfaktoren, Entzündungsbotenstoffe (Zytokine) und Immunzellen, aber auch lokale Regulatoren wie Sauerstoff und Nährstoffe sowie die Architektur des Gewebes oder Organs. In einer solchen Nische haben krankhaft veränderte Zellen ebenso wenig eine Chance, das Ruder an sich zu reißen, wie „Unkräuter“ in einem gut gepflegten, gedüngten und gewässerten Rasen.

Im jungen, gesunden Organismus gedeihen normale Zellen (Gras) wegen der Nährstoffversorgung (Wolke), Wachstumsfaktoren (Vogeldung) und der intakten Infrastruktur (Boden) so gut, dass sie Krebsvorstufen (Unkraut) in Schach halten. In alten oder geschwächten Organismen (rechts) herrscht ein anderer Selektionsdruck, sodass Krebsvorstufen nun einen Überlebensvorteil haben.
Eine Lebensweise und eine Umwelt, die unser Gewebe mit Karzinogenen wie Rauch oder UV-Licht oder Krankheitserregern in Kontakt bringt, kann dieses System ebenso aus dem Gleichgewicht bringen wie alle Vorgänge, die unseren Stoffwechsel oder Hormonhaushalt und unser Immunsystem stören – und das werden im Lauf der Lebensjahre immer mehr.
Irgendwann – meist im Alter – kippt das System; die Fitnesslandschaft verformt sich: Das umliegende Gewebe ist leicht entzündet und geschwächt; Barrieren halten nicht mehr dicht; der Hormonspiegel verändert sich und so weiter. Vereinzelte Krebsvorläuferzellen, die zwar längst alle nötigen Mutationen in sich versammelt haben, aber bisher ein Schattendasein führten, können unter den neuen Selektionsbedingungen die Oberhand gewinnen und zu großen Klonen heranwachsen. Voilà: Krebs.
Krankheiten, die erst gegen Ende oder nach unserer Reproduktionsphase zu Beeinträchtigungen oder zum Tode führen, sind für die natürliche Auslese unsichtbar. Vorher dagegen stabilisiert die Selektion die genetische Ausstattung von Zellen, die darauf optimiert sind, Krebsvorläufer in Schach zu halten. Diese stabilisierende Selektion ist bei großen, langlebigen Organismen wie Elefanten stärker als bei Mäusen: In ihnen treten zwar wegen der erheblich größeren Zahl von Stammzellen mehr potenziell krebsfördernde Mutationen auf, aber die betroffenen Zellen werden mit einer ebenfalls sehr viel größeren Wahrscheinlichkeit repariert oder ausgeschaltet, bevor etwas passiert. Sowohl Mäuse als auch Elefanten sterben in der freien Natur zumeist an etwas anderem, bevor sie Krebs bekommen können.
Jetzt kaufen – später zahlen!
Einige der Schutzmechanismen, die in jungen Jahren Krebs verhindern und den Fortpflanzungserfolg erhöhen, sind im Alter selbst am Verfall des Gewebes beteiligt und damit krebsfördernd. Diese doppelte, zunächst vorteilige, später nachteilige Wirkung einer genetischen Ausstattung nennt man „antagonistische Pleiotropie“. Ich komme im nächsten Artikel darauf zurück, denn einige unserer genetischen Anlagen für Autoimmunerkrankungen fallen vermutlich in diese Kategorie.
Ein Beispiel ist das Protein p53, das vom Tumorsuppressor-Gen TP53 codiert wird: Junge Mäuse schützt es vor Krebs, aber alte Mäuse erkranken dank seiner umso heftiger und schneller – offenbar, weil das Protein in altersbedingt leicht entzündetem Gewebe anders wirkt als in einem entzündungsfreien Umfeld, etwa indem es die Reparatur von DNA-Schäden behindert.
Kleine Nischen hemmen die Ausbreitung
Ob und wie schnell sich ein mutierter, nunmehr von der Selektion bevorteilter Klon im Gewebe durchsetzt, hängt unter anderem vom Verhältnis zwischen zwei evolutionären Kräften ab, nämlich der Gendrift (also der zufälligen Veränderung der Allelfrequenz innerhalb des Genpools) und der Selektion. Dieses Verhältnis wiederum wird durch die Größe und Struktur der Nischen geprägt, in denen teils nur kleine, teils sehr große Pools Platz finden.
So bilden fast alle Blut-Stammzellen einen einzigen, riesigen Pool von etwa 10.000 bis zu einigen 100.000 Zellen, die um eine knappe Ressource, nämlich um Plätze in den Überlebensnischen im Knochenmark konkurrieren. Unter diesen Bedingungen kann ein Klon mit einem kleinen Selektionsvorteil exponenziell anwachsen und sich schnell im ganzen Pool durchsetzen – so wie in einem großen Aquarium oder auf einem Kontinent mit entsprechend großer Population.
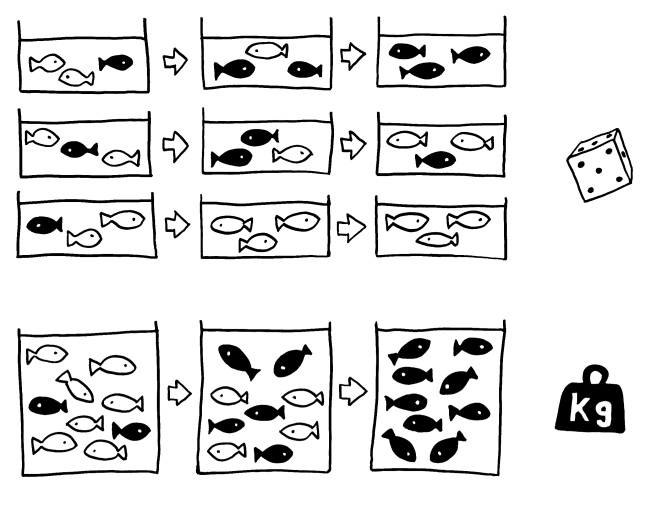
In kleinen Stammzellnischen wie im Darm kann die zufällige genetische Drift (Würfel) die Ausbreitung von Mutationen (schwarz) trotz Selektionsvorteil u. U. verhindern. In großen Stammzellpools wie den Blut-Stammzellen überwiegt der Selektionsdruck (Gewicht), sodass Mutationen, die den Zellen einen kleinen Vorteil bringen, sich fast immer durchsetzen.
Dagegen leben die Stammzellen unserer Darmschleimhaut in kleinen Nischen, den Krypten. Bei Mäusen enthalten diese Krypten nur jeweils 14 bis 20 Stammzellen. Auch wenn in einer Stammzelle Krebsvorläufer-Mutationen auftreten, die ihr einen Selektionsvorteil verschaffen, ist nicht gesagt, dass ihr Klon sich durchsetzt: Der Zufall kann ihn rechtzeitig auslöschen. Und selbst wenn dieselbe krebsfördernde Mutation auch in weiteren der zahlreichen Krypten auftritt, breiten sich die Krebsvorstadien dank der starken Untergliederung ihres Lebensraums nur mit linearer Geschwindigkeit aus. So bleibt genug Zeit, sie bei einem Screening zu entdecken und zu entfernen, bevor sie sich zu Darmkrebs auswachsen. Und auch ohne Screening kann es sein, dass man irgendwann an etwas anderem stirbt, bevor sich der Darmkrebs bemerkbar macht.
Das Gras kräftigen, nicht den Löwenzahn vergiften
Was aber bedeutet der ökologische und evolutionsbiologische Blick auf die Entstehung von Krebs für die Therapie? Sowohl Chemotherapien als auch Strahlen- oder Immuntherapien verändern das Ökosystem, in dem die Tumoren leben, und damit den Selektionsdruck auf die Krebszellen. Zellen, die einen Therapiezyklus überlebt haben, können sich zum einen anschließend sehr schnell erholen und wieder ausbreiten, da ihnen durch das Absterben der Konkurrenz mehr Ressourcen zu Gebote stehen. Und zum anderen kann die Therapie sowohl die Entstehung als auch die Ausbreitung therapieresistenter Zellklone fördern.
Viele Krebstherapien sind nämlich genotoxisch, führen also zu Veränderungen im Erbgut. Durch diese zusätzlichen Mutationen steigt das Risiko, dass Zellen neue Eigenschaften entwickeln, die sie gegen die Therapie resistent machen. Sie können beispielsweise Blockaden in ihren Signal-oder Stoffwechselwegen umgehen, die man durch die Verabreichung monoklonaler Antikörper erzeugt, um die Tumoren „auszuhungern“ oder zumindest ihr Wachstum zu hemmen. Und da eine solche Resistenz einen denkbar großen Überlebensvorteil mit sich bringt, kann es sehr schnell zu Rezidiven kommen, also zum Auftauchen neuer, nunmehr kaum noch zu bekämpfender Tumoren.
Zur Zeit werden im Tierversuch neue Therapieansätze erprobt, die diese fatale Wirkung vieler Krebstherapien vermeiden sollen. Sie laufen der Intuition zuwider, denn sie zielen nicht darauf ab, alle Tumorzellen abzutöten, sondern versuchen, die weniger aggressiven Tumorzellen gegen die aggressiveren auszuspielen:
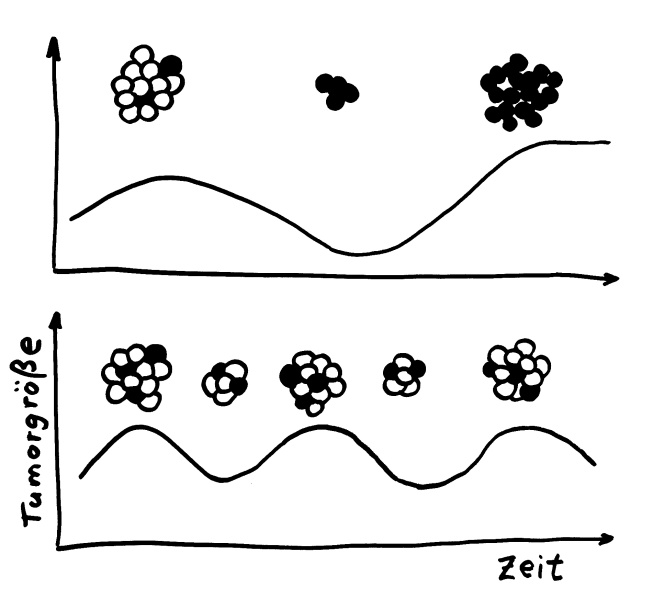
Oben: Herkömmliche Krebstherapien zielen auf ein vollständiges Absterben des Tumors ab und bringen oft resistente Zellen (schwarz) hervor, die sich schnell vermehren. – Unten: Zur Zeit werden neue Ansätze mit niedrigen Chemo-Dosen erprobt, die die weniger schädlichen Zellen im Tumor (weiß) nicht komplett abtöten, sodass diese die schädlicheren Zellen (schwarz) in Schach halten. Krebs soll so zu einer chronischen, beherrschbaren Erkrankung gemacht werden.
Eine solche Therapie ist nicht zytotoxisch, sondern zytostatisch. Sie hemmt also die Zellteilung und reduziert damit das Risiko weiterer Treibermutationen. Krebs soll auf diese Weise in eine beherrschbare chronische Erkrankung verwandelt werden, mit der der Organismus noch lange leben kann. Bei Mäusen funktioniert das schon ganz gut.
Ein zweiter Ansatz ist ebenso kontraintuitiv: Seit Jahrzehnten versucht man, Tumoren auszuhungern, indem man ihre Fähigkeit blockiert, im umliegenden Gewebe die Entstehung neuer Blutgefäße anzuregen. Denn über diese Blutgefäße wird der Tumor ja mit Ressourcen versorgt, die ihn zum weiteren Wachstum befähigen. Dummerweise fördert man mit solchen Therapien auch die Metastasierung, die viele Krebsarten erst tödlich macht: Der Sauerstoffmangel im Inneren von Primärtumoren, die nicht hinreichend durch Blutgefäße versorgt werden, ist ein Selektionsdruck, der beweglichen Krebszellen einen immensen Vorteil verschafft, denn sie können den Primärtumor verlassen und sich an anderer Stelle ansiedeln. Im Tierversuch hat es sich als vielversprechend erwiesen, Primärtumoren stattdessen gut mit Sauerstoff zu versorgen, um keinen Anreiz zur Durchsetzung Metastase-fähiger Zellklone zu setzen.
Um das ganz deutlich zu sagen: Nichts davon ist auch nur annähernd marktreif. Es kann sein, dass diese neuen Ansätze im Sande verlaufen, wie so viele vermeintliche Innovationen in den letzten Jahrzehnten, die den Schritt von der Maus zum Menschen nicht geschafft haben oder den Teufel mit dem Beelzebub austreiben, sodass die Patienten an schrecklichen Nebenwirkungen sterben. Sie werden auch sicher nicht gegen alle Arten von Krebs gleichermaßen wirken.
Die Therapien interessieren mich auch weniger als die Konzepte, auf denen sie fußen. Vor allem geht es mir natürlich um die Schnittmenge zwischen Onkologie und Immunologie – etwa um die These, dass bestimmte Autoimmunerkrankungen im Grunde erfolgreiche, aber aus dem Ruder gelaufene Krebsbekämpfungsmaßnahmen unseres Körpers sind. Dazu im nächsten Artikel mehr.
Literatur
M. Casás-Selves, J. DeGregori (2011): How cancer shapes evolution, and how evolution shapes cancer
D. Hanahan, R. A. Weinberg (2011): Hallmarks of Cancer: The Next Generation
M. Greaves, C. C. Maley (2012): Clonal evolution in cancer
A. I. Rozhok, J. DeGregori (2015): Toward an evolutionary model of cancer: Considering the mechanisms that govern the fate of somatic mutations
Pressemitteilung dazu: Evolution, not just mutation, drives development of cancer