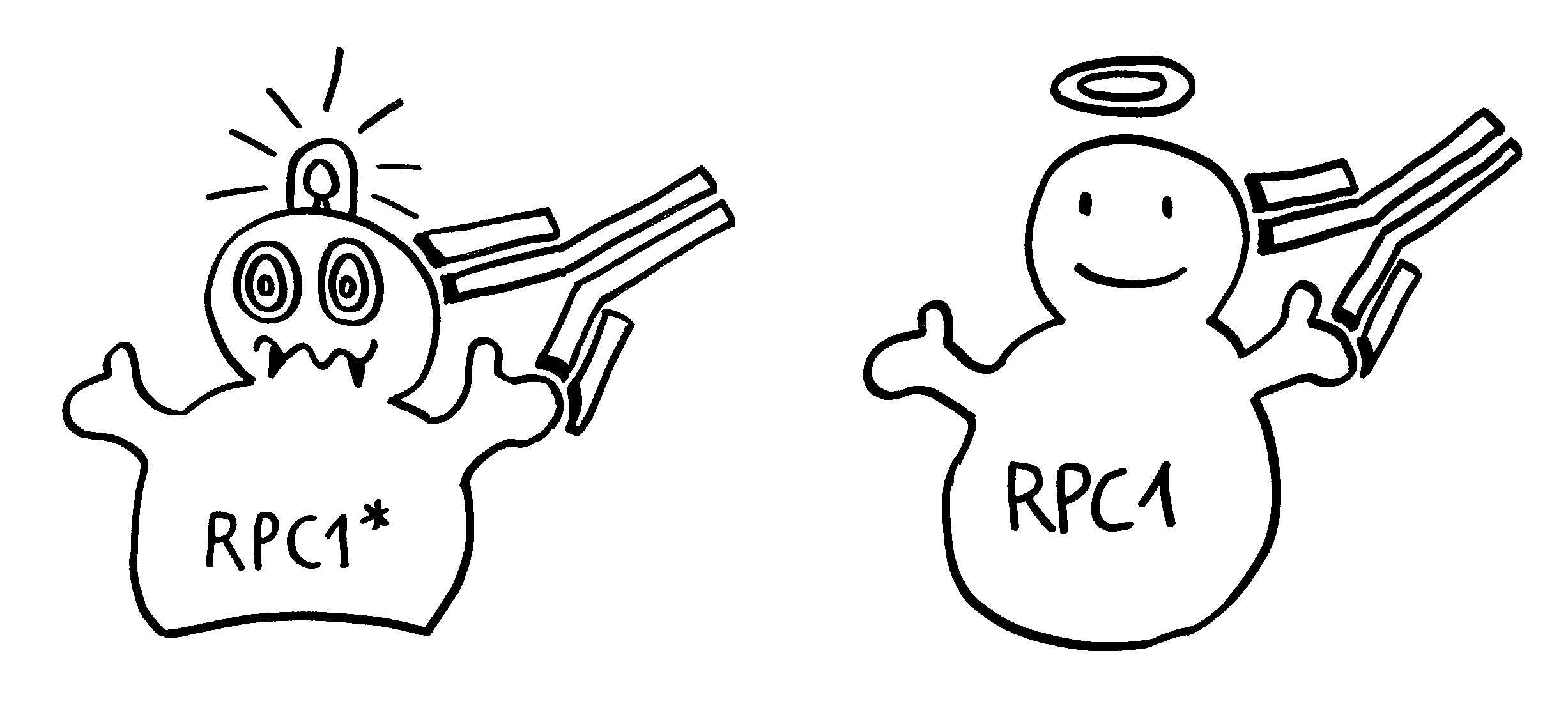
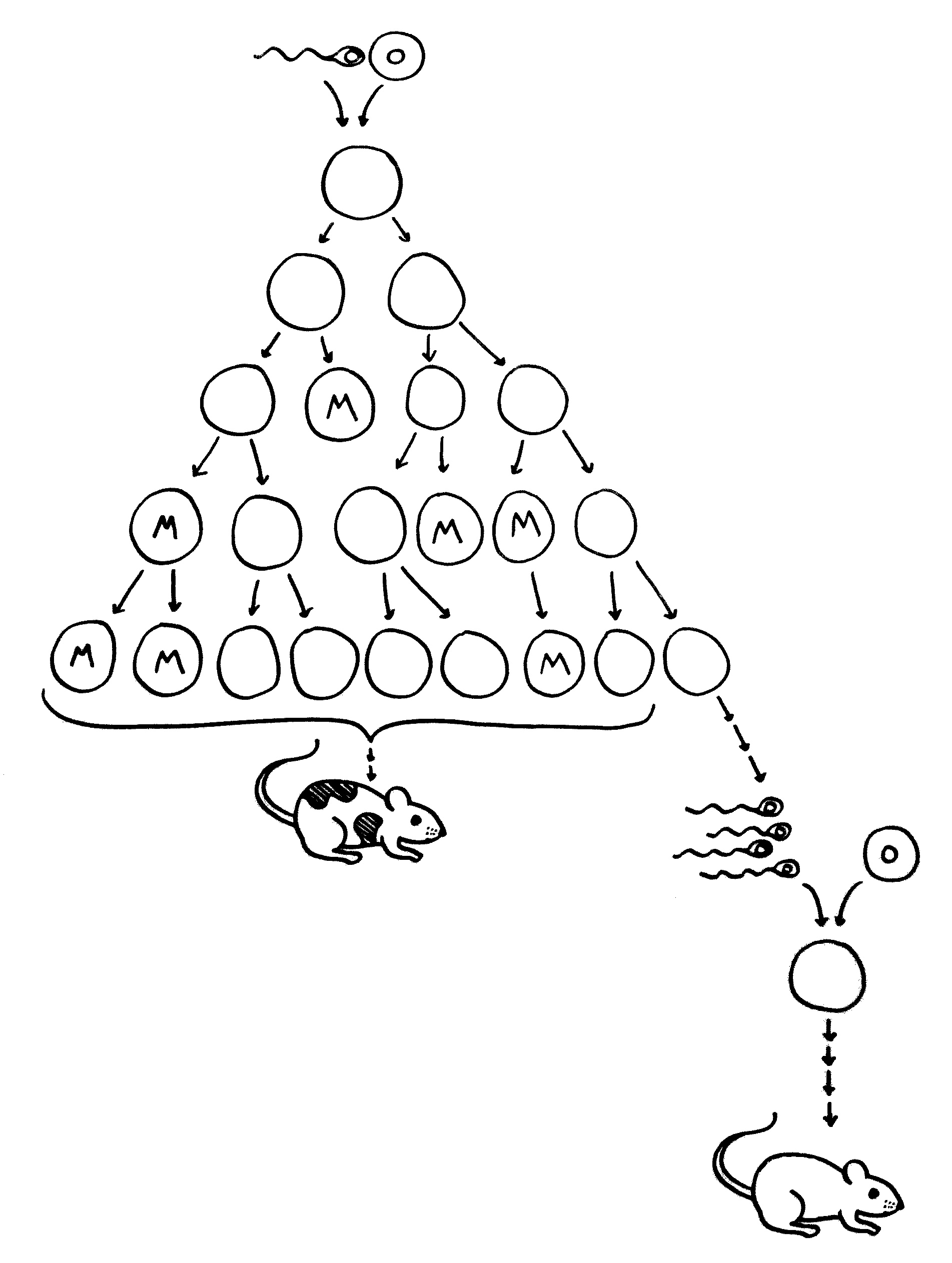
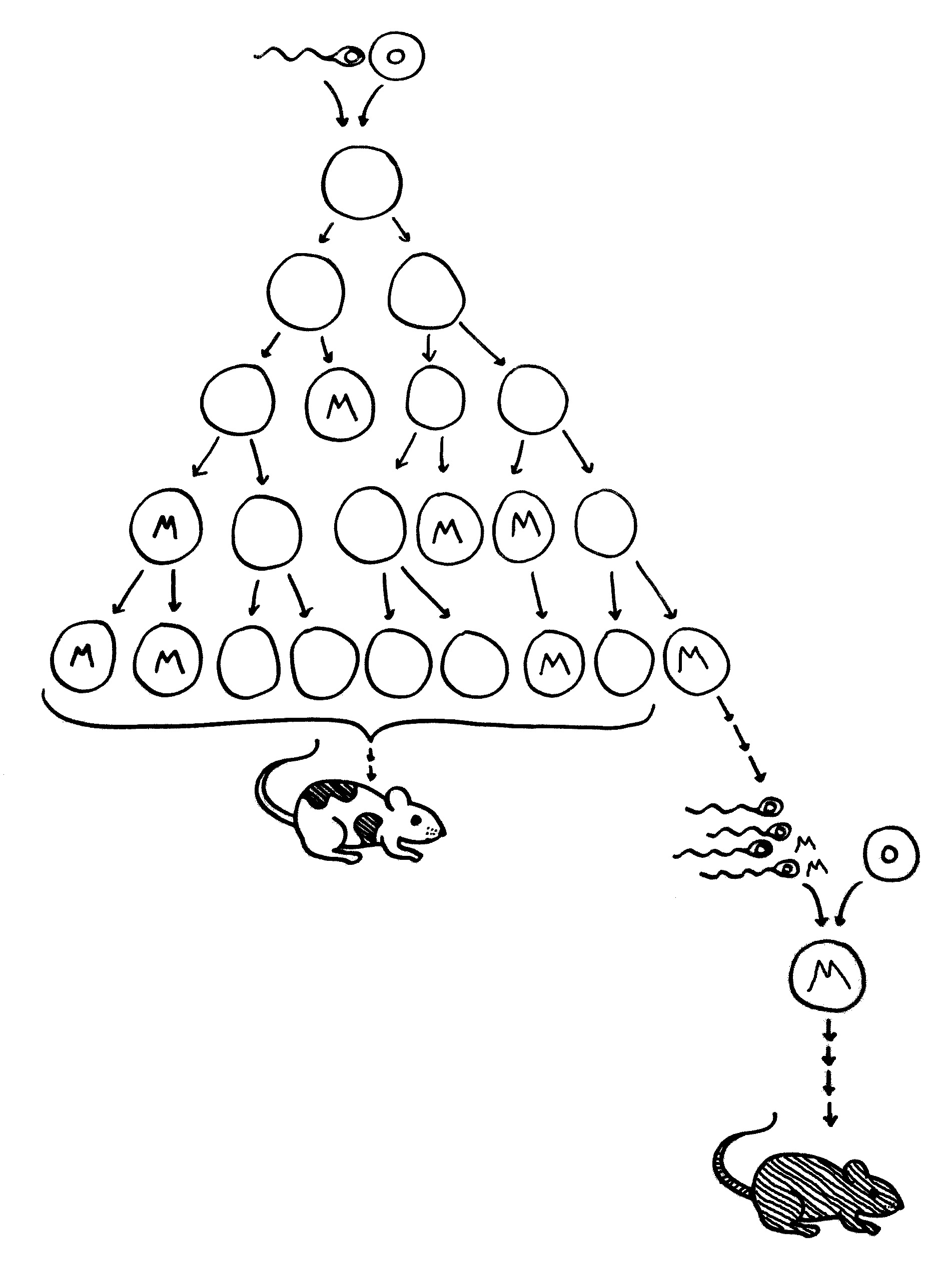
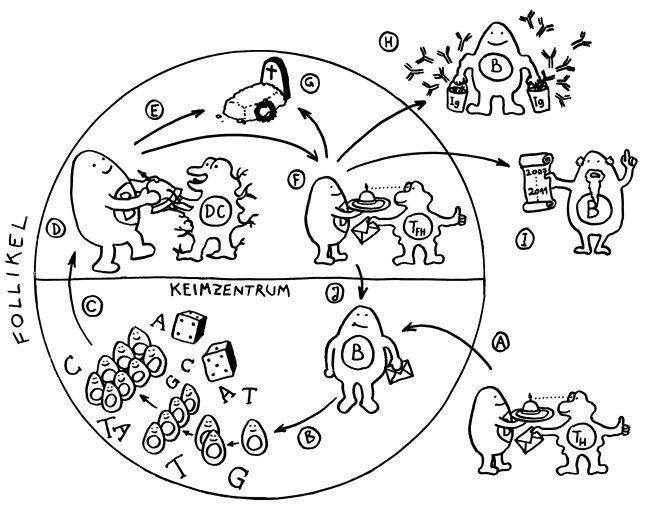
1. Eine Zelle wird zur Krebsvorläuferzelle; sie produziert sehr viel von einem für unreife Zellen
typischen Protein.
2. Eine Mutation (MUT) in einer solchen Zelle verändert das Protein.
3. Im Tumor kommen Zellen mit der Mutation und solche mit dem normalen Protein vor, dem
sogenannten Wildtyp (WT).
4. Aus mutierten Zellen wird das veränderte Protein freigesetzt, zum Beispiel, wenn sie sterben.
5. Antigenpräsentierende Zellen nehmen dieses Antigen auf und präsentieren es zusammen mit Kostimulationssignalen (Kerze).
6. Das Antigen wird wegen seiner Fremdartigkeit als gefährlich eingestuft und aktiviert das Immunsystem.
7. Die aktivierten Effektorzellen bekämpfen den Tumor. Dabei treten weitere Proteine aus – sowohl veränderte als auch unveränderte.
8. Auch das normale Protein wird nun als Antigen präsentiert, zusammen mit Kostimulationssignalen.
9. Im Kontext der laufenden Immunreaktion wird auch das normale Autoantigen als gefährlich eingestuft (molekulare Mimikry); autoreaktive Lymphozyten werden aktiviert (bystander activation).
10. Fernab vom Tumor, zum Beispiel in Blutgefäßwänden, produzieren unreife Zellen dasselbe Antigen und werden damit zum Ziel der Abwehr.
11. Die Lymphozyten greifen die unreifen Zellen an und setzen so noch mehr der Autoantigene frei, auf die sie reagieren.
12. Dieser Teufelskreis läuft auch weiter, wenn der Tumor längst verschwunden ist: Die Autoimmunerkrankung hat sich etabliert.
Sie dürfen diese Zeichnung gerne in Folien etc. übernehmen, sofern Sie die Quelle angeben: Dr. Andrea Kamphuis, https://autoimmunbuch.de