Kaum ein halbes Jahr nach der Verleihung des Nobelpreises an Yoshinori Ōsumi, der diesen zellulären Schutzmechanismus an Hefen erforscht hat, schreibe ich nun auch was zur Autophagie. Den Schwerpunkt lege ich dabei auf die Berührungspunkte zu meinem eigentlichen Thema, den Autoimmunerkrankungen.
Regelmäßige Müllabfuhr
In unseren Zellen ist ständig Müll zu entsorgen, auch wenn sie bei bester Gesundheit sind: Überraschend viele frisch hergestellte Proteine sind falsch zusammengefaltet, und wenn das nicht sofort korrigiert werden kann, müssen sie wieder abgebaut werden. Andere Makromoleküle werden mit der Zeit beschädigt oder einfach nicht mehr benötigt. Sogar ganze Organellen wie Mitochondrien müssen zerlegt werden, um nicht die Zelle zu verstopfen, leck zu schlagen oder schädliche biochemische Reaktionen in Gang zu setzen. Nicht nur, dass das alte Material schaden kann: Es besteht auch aus wertvollen Bausteinen wie Aminosäuren, die die Zelle recyceln muss, um mit ihrem begrenzten Energievorrat auszukommen.
Schienentransport
Proteine können auf zwei Wegen abgebaut werden. Etwa 80 Prozent werden in tonnenförmigen Strukturen zerlegt, den Proteasomen. Große Proteinkomplexe und Organellen passen allerdings nicht in diese Abfalltonnen hinein. Sie werden stattdessen von Lysosomen verdaut, von Membranbläschen, die mit Enzymen gefüllt sind: den sauren Hydrolasen. Die Lysosomen halten sich in der Nähe des Zellkerns auf und verschmelzen dort mit sogenannten Autophagosomen. Diese sind von einer doppelten Membranschicht umgeben und enthalten den abzubauenden Zellmüll. Sie entstehen überall in der Zelle, wo es etwas zu entsorgen gibt: Dort bilden sich zunächst Doppelmembran-Lappen, die Phagophoren, die sich dann um den Schrott herumschmiegen und zu einem Müllsack verschmelzen.
Die verschlossenen Müllsäcke (die Autophagosomen) werden dann an Schienen – den röhrenförmigen Mikrotubuli, die zum sogenannten Zellskelett gehören – zu den Lysosomen transportiert. Die kleinen Lysosomen vereinigen sich mit den größeren Autophagosomen zu Autolysosomen, sodass die Enzyme den Zellmüll zu kleinen Bausteinen zerlegen können. Diese verlassen die Autolysosomen und stehen im Zytoplasma zum Aufbau neuer Makromoleküle zur Verfügung.
Neben dem allgemeinen Recycling gibt es auch eine selektive Sondermüllabfuhr: Bei Bedarf werden zum Beispiel Mitochondrien, Fetttröpfchen, Ribosomen oder in die Zelle eingedrungene Pathogene mit speziellen Markern (sozusagen Abholzetteln) gekennzeichnet und daraufhin gezielt entsorgt.
Bedarfsgesteuerter Abfuhrtakt
Den ganzen Vorgang nennt man Makroautophagie oder kurz Autophagie, weil die Zelle Teile ihrer selbst (griechisch auto) vertilgt (griechisch phagein = essen). Die daran beteiligten Proteine sind in den Atg-Genen codiert (autophagy-related genes), von denen man in der Hefe allein 36 identifiziert hat. Bei Säugetieren wie dem Menschen sind von vielen der Proteine zudem mehrere Varianten bekannt, sogenannte Isoformen.
Wenn der Prozess gestört ist, beispielsweise weil eines dieser Gene einen Defekt hat, gerät die Zelle aus dem Gleichgewicht, aus der Homöostase: Sowohl überzogenes Reinemachen als auch ein Streik der Müllabfuhr können zu Funktionsstörungen oder zum Zelltod führen.
Es gibt jedoch Situationen, in denen der Abfuhrtakt erhöht werden muss – etwa wenn die Zelle hungert und unnötige Strukturen abbauen muss, um Lebenswichtiges damit herzustellen oder zu erhalten, oder wenn Stress wie zum Beispiel eine Infektion, Chemikalien oder UV-Licht den Verschleiß von Zellkomponenten beschleunigen. Auch Bakterien und andere Pathogene sowie ihre Überreste werden bei einer Infektion per Autophagie aus dem Zytoplasma beseitigt.
Dem Tod von der Schippe springen
Wie am Ende meiner Todesarten-Übersicht erläutert, konkurrieren am Anfang der Zelltodesprogramme Überlebenssignale mit Sterbesignalen. Ist eine Zelle nicht tödlich verwundet, unrettbar infiziert oder dermaßen entgleist, dass ihr Fortbestand andere Zellen gefährden würde, kann das Ruder noch herumgerissen werden: Dann wird die Autophagie verstärkt, um die Zelle ins Gleichgewicht zurückzubringen, und zugleich das Sterbeprogramm blockiert.
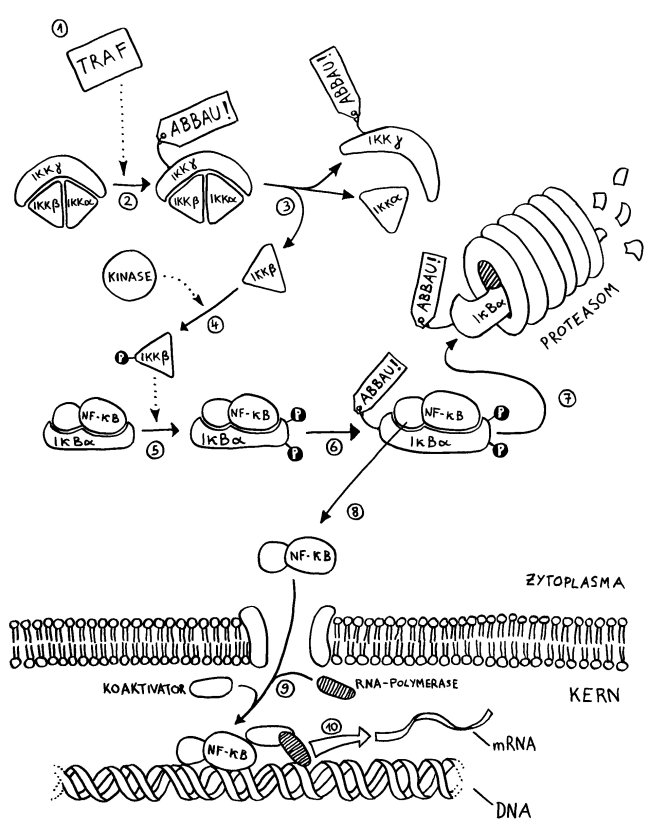
Zu den lebenserhaltenden Signalen zählen etwa der Kontakt zur extrazellulären Matrix, bestimmte Hormone, Chemokine oder Neurotransmitter sowie dezidierte Überlebensfaktoren (survival factors) wie IGF-1 (insulin-like growth factor 1). Sie binden an Rezeptoren auf der Zelloberfläche, woraufhin an der Innenseite der Zellmembran Signalketten in Gang kommen, die zur Bindung des Transkriptionsfaktors NF-κB an bestimmte Gene im Zellkern führen. Diese anti-apoptotischen Gene (zum Beispiel Bcl-2) werden dann abgelesen, und die Genprodukte hemmen die Todesprogramme der Zelle.
Das kann beispielsweise durch eine verstärkte Autophagie von Mitochondrien geschehen, die bei Zellstress beschädigt wurden und daraufhin Cytochrom C freisetzen, das die Effektor-Phase einer intrinsischen Apoptose einleitet: Kommen die Autophagosomen dem zuvor, bleibt die Zelle am Leben.
Schädliche Durchhalteparolen
Bei mildem Zellstress oder entsprechend konditionierten, also an erhöhte Stresslevel gewöhnten Zellen können Überlebenssignale also reichen, um beispielsweise eine Apoptose zu verhindern. Ist der Stress dagegen zu stark, überwiegen die pro-apoptotischen Signale, und die Zelle stirbt.
Dabei ist ein Überleben nicht immer gut für den Organismus und ein Zelltod nicht immer schlecht. Man denke nur an Krebszellen im Inneren eine Tumors, die ständig Stress wie Sauerstoff- oder Nährstoffmangel erfahren und sich trotzdem weigern einzugehen: Dank genetischer Veränderungen können sie die Apoptose verhindern, obwohl sie aus ihrer Umgebung eigentlich mehr Sterbekommandos als Überlebenssignale empfangen.
Und um allmählich die Kurve zu den Autoimmunerkrankungen zu kriegen: Autoreaktive T- und B-Zellen können nur deshalb dauerhaft Unheil anrichten, weil sie entweder in sogenannten Überlebensnischen wie dem Knochenmark mit Überlebenssignalen versorgt werden – oder weil sie, wie Krebszellen, ihr Überleben von solchen Signalen unabhängig gemacht haben.
Autophagie und Autoimmunerkrankungen
Autophagie ist im Immunsystem besonders wichtig, weil Immunzellen sich bei Bedarf in kurzer Zeit massiv vermehren müssen und ihr Stoffwechsel nach ihrer Aktivierung Hochleistungen vollbringen muss, für die viel Energie benötigt wird.
Genomweite Assoziationsstudien (GWAS) haben ergeben, dass etliche kleine Varianten in den Autophagie-Genen, vor allem Atg5 und Atg16l1, mit der Neigung zu Lupus (SLE), Morbus Crohn, Multipler Sklerose und rheumatoider Arthritis assoziiert sind. Sowohl ein Übermaß als auch ein Mangel an Autophagie kann zu Autoimmunstörungen beitragen, je nachdem, in welchen Zellen die Müllabfuhr aus dem Lot gerät.
Über welche Mechanismen eine genetisch angelegte Autophagie-Fehlfunktion zu den Erkrankungen beiträgt, ist nicht immer klar. Im Folgenden führe ich einige mögliche Mechanismen an, die zum Teil erst in Tiermodellen und Gewebekulturen untersucht wurden. Der Weg zu einer Therapie ist also noch lang, und wie so oft wird die Kunst darin bestehen, den Teufel nicht mit dem Beelzebub auszutreiben – also die Autophagie gezielt in bestimmten Zelltypen anzukurbeln oder zu bremsen, ohne dass es andernorts zu unerwünschten Effekten kommt.
Autophagie-Defizienz in der Entwicklung von Zellen der erworbenen Abwehr
T-Zellen entstehen im Knochenmark und reifen im Thymus heran, wo sie eine starke Selektion durchlaufen, die für die Selbsttoleranz des Immunsystems sorgt. Die dafür verantwortlichen Thymus-Epithelzellen (TEC) betreiben viel Autophagie, um den jungen T-Zellen ständig neue Antigene auf ihren MHC-Komplexen präsentieren zu können. Mäuse, deren TEC wegen einer Atg5-Defizienz zu wenig Material recyceln, entwickeln starke Autoimmunerkrankungen, da die autoreaktiven unter den T-Zellen im Thymus nicht zuverlässig aussortiert werden.
In den jungen T-Zellen selbst scheint Autophagie keine besondere Rolle zu spielen – wohl aber bei der Entstehung frischer B-Zellen: Bei einer Atg5-Defizienz sterben ihre Vorläufer ab.
Autophagie-Störungen in reifen Zellen der erworbenen Abwehr
Die T- und B-Zellen von Lupus-Patienten scheinen mehr Autophagie zu betreiben als die gesunder Menschen, und Wirkstoffe, die diese Überaktivität hemmen, scheinen sowohl Mäusen als auch Menschen mit Lupus gutzutun.
Aber zu wenig Autophagie schadet ebenfalls: Autophagie-Defekte führen in T-Zellen zur Anhäufung von überalterten Mitochondrien, die unter anderem reaktionsfreudigen Sauerstoff (ROS) freisetzen, was die T-Zellen vorzeitig absterben lässt. Auch fehlen den T-Zellen wegen ihrer schlechten Recycling-Quote ATP und Rohstoffe, sodass sie sich kaum noch aktivieren und zur Vermehrung anregen lassen.
Vorzeitig alternde T-Zellen mit Energiedefiziten wurden zum Beispiel im Blut von Menschen mit rheumatoider Arthritis gefunden. Man vermutet, dass der so entstehende Mangel an funktionstüchtigen T-Zellen (Lymphopenie) durch eine übermäßige Vermehrung autoreaktiver T-Zellen kompensiert wird.
Autophagie-Defizienz in Zellen der angeborenen Abwehr
Bei der Beseitigung apoptotischer Zellen durch Makrophagen sind auf beiden Seiten Autophagie-Proteine im Spiel: Die sterbenden Zellen brauchen sie, um „Kommt und holt mich“-Signale auszusenden, und in den Fresszellen sind sie an der Verdauung der vertilgten Zellbestandteile beteiligt. Bei einer Autophagie-Defizienz können sich tote Zellen oder Zellbestandteile im Gewebe ansammeln, wie man es beispielsweise bei Lupus beobachtet hat. Aus nicht rechtzeitig entsorgten toten Zellen treten Autoantigene wie DNA oder RNA aus, die Entzündungen und Autoimmunreaktionen auslösen können.
Vor allem Makrophagen und Neutrophile bilden, wenn sie entsprechend aktiviert werden, Inflammasomen: Proteinkomplexe, die für die massenhafte Ausschüttung der Zytokine IL-1β oder IL-18 sorgen, die wiederum T- und B-Zellen anlocken. Eine intakte Autophagie bremst die Bildung von Inflammasomen, denn bei guter Verdauung brauchen die Zellen keine Hilfe von außen. Schwächelt die Autophagie in Makrophagen oder Neutrophilen, kann es aber zu überzogenen Reaktionen der erworbenen Abwehr auf harmlose Antigene kommen.
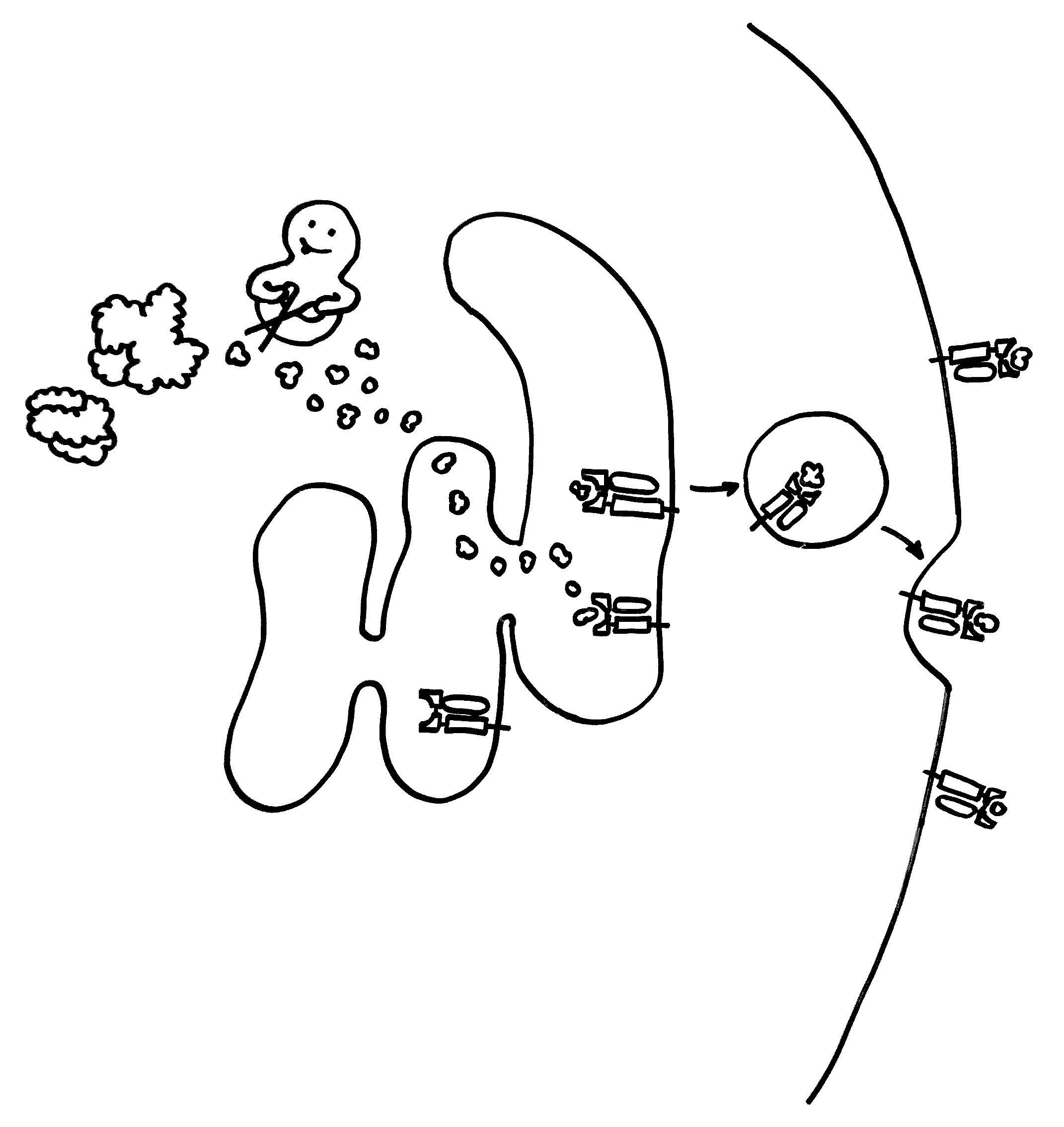
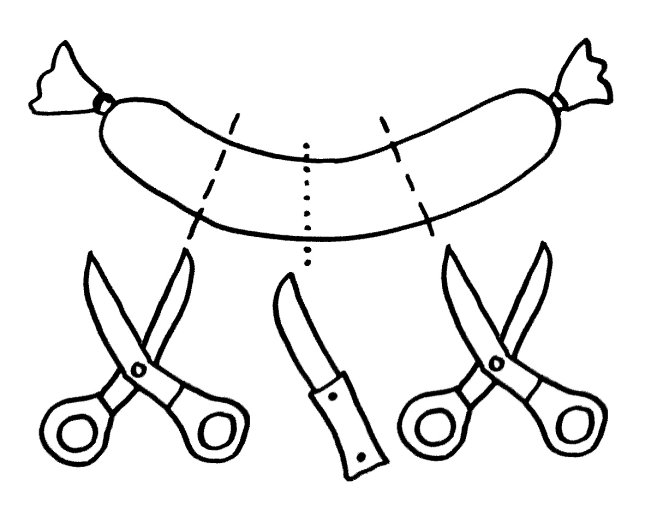
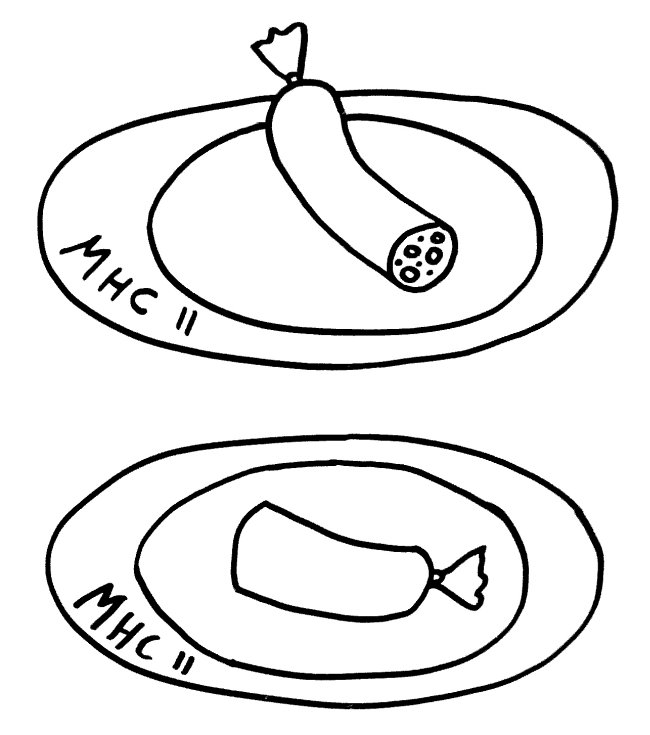
Auch die Antigen-Präsentation auf MHC-Klasse-II-Komplexen gelingt Makrophagen und dendritischen Zellen offenbar nur mithilfe von Autophagie, denn die aufgenommenen Antigene müssen ja zerschnitten, zu den MHC-Klasse-II-Komplexen (den „Präsentiertellern“) transportiert und nach dem Vorzeigen wieder nach innen geschafft und entsorgt werden. Autophagosomen können entweder (bei der Autophagie) mit Lysosomen fusionieren oder (zum Zwecke der Antigen-Präsentation) mit den Kompartimenten, in denen die MHC-Klasse-II-Komplexe auf ihre Beladung mit Antigenen warten. Vielleicht führen bestimmte Genvarianten dazu, dass die Autophagosomen ihren Inhalt sozusagen an die falsche dieser beiden Adressen liefern. So können Varianten im Gen Atg16l1 das Morbus-Crohn-Risiko erhöhen – vermutlich weil die T-Zellen dann nicht genug Pathogen-Antigene präsentiert bekommen und daher nicht richtig aktiviert werden.
Bei rheumatoider Arthritis präsentieren Makrophagen und dendritische Zellen den T-Zellen chemisch leicht veränderte, sogenannte citrullinierte Antigene. Man vermutet, dass diese Veränderungen die Selbsttoleranz des Immunsystems aushebeln und zu den Autoimmunreaktionen in dem Gelenken führen. Hemmt man die Autophagie in den antigenpräsentierenden Zellen, werden vor allem weniger citrullinierte Antigene präsentiert.
Autophagie-Störungen in anderen Zellen
An Morbus Crohn, einer chronischen Entzündung, die bei Menschen mit entsprechender genetischer Disposition von Keimen aus der Darmflora ausgelöst oder zumindest beeinflusst wird, kann ein Versagen der Autophagie in den Zellen der Darmschleimhaut beteiligt sein: Bestimmte Escherichia-coli-Stämme dringen in die Epithelzellen ein, um sich in ihrem Inneren zu vermehren. Eine intakte Autophagie verhindert oder begrenzt diese Vermehrung; bei Autophagie-Defekten können die Keime dagegen überhand nehmen und tiefer ins Gewebe eindringen, wo sie die chronische Entzündung auslösen.
Störungen der internen Müllabfuhr könnten in allen möglichen Zellen auch das Endoplasmatische Reticulum (ER) unter Stress setzen, ein membranreiches, netzförmiges Organell, das unter anderem der Protein-Herstellung dient. ER-Stress wiederum trägt bei Typ-2-Diabetikern zur Insulinresistenz bei, und zwar auf mehreren Wegen: Er stört die Insulin-Herstellung in den Betazellen der Bauchspeicheldrüse, treibt diese Zellen zur Apoptose, löst Entzündungen aus und führt zu einer Anhäufung von Fetten.
Auch bei Hashimoto-Thyreoiditis scheint die Müllabfuhr zu streiken: In den Follikel-Epithelzellen der Schilddrüse von Menschen mit dieser Erkrankung hemmt hoch dosiertes Jod die Autophagie und kurbelt zugleich die Apoptose an – zumindest in vitro. Sollte sich das in vivo bestätigen, könnte man versuchen, die schwächelnde Autophagie in den Schilddrüsenzellen zu stärken und so die Organzerstörung aufzuhalten.
Literatur:
Abhisek Bhattacharya, N. Tony Eissa (2013): Autophagy and autoimmunity crosstalks
Liam Portt et al. (2011): Anti-apoptosis and cell survival: A review
Chengcheng Xu (2016): Excess iodine promotes apoptosis of thyroid follicular epithelial cells by inducing autophagy suppression and is associated with hashimoto thyroiditis disease (Abstract)
Zhen Yang et al. (2015): Autophagy in autoimmune disease