Im vorigen Beitrag ging es um Polly Matzingers danger theory, der zufolge ein Gewebe, das eine Infektion oder einen physikalisch oder chemisch bedingten Schaden erleidet, Gefahren- oder Schadsignale aussendet, die das Immunsystem aktivieren. Solche DAMPs (danger- oder damage-associated molecular patterns) können durch Traumata wie Quetschungen, Verschleiß, Verbrennungen oder Infarkte aus dem Inneren von Zellen austreten, in denen sie sich normalerweise verbergen, und lösen dann im Immunsystem eine sogenannte sterile Entzündung aus. Diese dient der Beseitigung verletzter und toter Zellen und der anschließenden Gewebsreparatur.
Sterile Entzündungen haben vieles mit Entzündungen gemeinsam, die durch Infektionen ausgelöst werden; beispielsweise erkennen dieselben Rezeptoren auf antigenpräsentierenden Zellen (dendritischen Zellen und Makrophagen) sowohl DAMPs als auch PAMPs, also pathogen-associated molecular patterns. Eine sterile Entzündung belastet aber das Gewebe weniger und läuft oftmals ohne volle Einschaltung der erworbenen Abwehr ab: Der Einsatz von antigenspezifischen T- und B-Zellen ist ja nicht nötig, wenn die Schadensursache kein Pathogen ist, das diese Zellen passgenau erkennen, bekämpfen und erinnern müssten.
DAMPs und Autoimmunerkrankungen
Sterile Entzündungen und DAMPs interessieren uns hier, weil sie mit Autoimmunerkrankungen in Verbindung gebracht wurden: Nur bei sehr wenigen dieser Erkrankungen ist eine Infektion als Auslöser nachgewiesen; bei anderen wie Rheuma ist die Beteiligung von Pathogenen umstritten. Bei vielen scheint das Immunsystem aber ganz ohne Mitwirkung eines Keims aus dem Ruder zu laufen und sich gegen körpereigene Strukturen zu richten – und zwar dauerhaft, sofern die angeborene Abwehr doch die erworbene Abwehr zu Hilfe ruft.
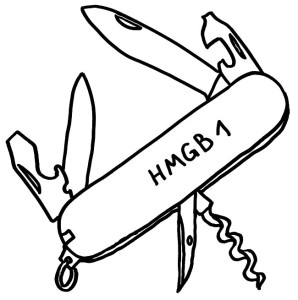
Die Auslösung steriler Entzündungen ist nur eine der vielen Funktionen von HMGB1.
Ein wichtiges Alarmsignal mit dem sperrigen Namen HMGB1 spielt offenbar bei der Pathogenese von Lupus (SLE), rheumatoider Arthritis, Sjögren-Syndrom, Vaskulitiden wie Morbus Behçet, Sklerodermie, Typ-1-Diabetes und weiteren Krankheiten eine unglückliche Rolle: Im erkrankten Gewebe oder im Blut der Betroffenen ist seine Konzentration gegenüber Gesunden stark erhöht, und in Tierversuchen lassen sich einige Autoimmunreaktionen durch die Ausschaltung von HMGB1 bremsen.
Gerade wegen seiner Schlüsselrolle beim Anstoßen von Entzündungsreaktionen reguliert unser Körper die örtliche Konzentration und das Aktivitätslevel dieses Moleküls normalerweise strikt, und zwar auf mehreren Wegen. Um diese raffinierte Selbstkontrolle – die checks and balances, die bei Autoimmunerkrankungen offenbar versagen – soll es im Folgenden gehen. Um sie zu verstehen, müssen wir uns zunächst das Molekül genauer ansehen.
Steckbrief
Das 1973 aus Kalbsbries (Thymus) isolierte Protein heißt ausgeschrieben „High mobility group box 1“, weil es sich auf einem Elektrophorese-Gel schnell bewegt. Da diese Eigenschaft weiter nichts zur Sache tut, bleiben wir im Folgenden bei der Abkürzung. Das 215 Aminosäuren lange Protein ist evolutionär stark konserviert; 99 Prozent der Aminosäuren stimmen zwischen Maus und Mensch überein. Normalerweise hält es sich im Zellkern auf. 1999 entdeckte man, dass es nicht nur im Zellinneren vorkommt, sondern insbesondere von Makrophagen auch ausgeschieden wird – und zwar relativ spät in einem Entzündungsprozess, nämlich frühestens acht Stunden nach Aktivierung der Immunzellen. So lange brauchen die Zellen, um das Molekül so zu modifizieren, dass es ausgeschleust werden kann.
Für diese Ortsverlagerung und den damit einhergehenden Funktionswechsel sind zwei in der folgenden Abbildung mit „Kerntransport“ bezeichnete Abschnitte zuständig, in denen die Aminosäure Lysin angereichert ist. Diese Lysine werden bei Bedarf acetyliert oder phosphoryliert; ihnen werden also Acetyl- oder Phosphatgruppen angehängt. Dank dieses Signals werden sie aus dem Zellkern, in dem sie sonst an die DNA gebunden sind, ins Zytoplasma und ggf. ganz aus der Zelle transportiert.
Das Molekül gliedert sich in drei große Funktionseinheiten: zwei sogenannte HMG-Boxen (A und B), mit denen es sich relativ locker und nicht sequenzspezifisch an DNA anlagert, sowie einen sogenannten C-Schwanz, der bei der Krümmung von DNA zum Einsatz kommt, also etwa bei der Reparatur der Doppelhelix oder beim Ablesen (der Transkription) von Genen. Box B enthält eine Bindungsstelle für den wichtigen Zelloberflächen-Rezeptor TLR4 und an ihrem Ende eine Bindungsstelle für den ebenfalls an Zelloberflächen angesiedelten Rezeptor RAGE. Von beiden wird noch die Rede sein. Weitere Bindungsstellen sind bekannt, hier aber nicht eingezeichnet.
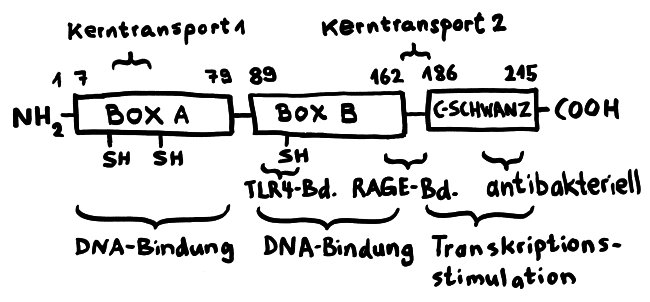
Wichtig sind noch drei Positionen, die mit der Aminosäure Cystein besetzt sind. Deren Seitenketten oder „Reste“ sind – zumindest solange sich das Molekül im Zellkern aufhält – sogenannten Thiolgruppen (-SH), die aus einem Schwefel- und einem Wasserstoffatom bestehen. Ich komme weiter unten auf sie zurück, denn ihre chemische Veränderung bestimmt die Aktivität des Moleküls nach seiner Ausschleusung aus der Zelle.
Je nach Aufenthaltsort unterschiedliche Aufgaben
HMGB1 ist weit mehr als ein DAMP, ein Alarmsignal für antigenpräsentierende Zellen: Außerhalb des Immunsystems übt es zum Beispiel lebenswichtige Funktionen im Nervensystem und im Stoffwechsel aus.
Normalerweise hält es sich im Zellkern auf, wo es als DNA-Chaperon, also als Faltungs- oder Krümmungshelfer für die Doppelhelix dient: Es stabilisiert die dreidimensionale Struktur der Nukleosomen, und es fördert die Transkription der Gene, die somatische Rekombination in B- und T-Zellen, die Reparatur von DNA-Schäden und den Erhalt der Telomere – der Schutzkappen an den Enden der Chromosomen. Kurz: Ohne HMGB1 keine intakten Chromosomen.
Wird es aus dem Zellkern ins Zytoplasma transportiert, was bei Zellstress und vor allem bei einer bakteriellen Infektion der Zelle passiert, fördert es die sogenannte Autophagie, eine partielle Selbstverdauung der Zelle, die den Bakterien die Lebensgrundlage entzieht. Zugleich hemmt es die Apoptose, also den kontrollierten Zelltod.
In die extrazelluläre Flüssigkeit gelangt es entweder durch passive Freisetzung aus allen möglichen beschädigten, sterbenden oder toten Zellen – dann ist es ein Alarmsignal, ein DAMP – oder durch aktive Ausschüttung aus Immunzellen, vor allem aus antigenpräsentierenden Zellen, also Makrophagen und dendritischen Zellen – dann kann man es als Zytokin auffassen, also als Immunsystem-internen Botenstoff.
Es kann als Chemokin dienen, also als Lockstoff für Immunzellen. Es kann andere Immunzellen aktivieren oder deaktivieren, zur Vermehrung bringen und zur Produktion und Ausschüttung von Boten- oder Wirkstoffen anhalten. Es kann alleine agieren, sich aber auch mit anderen Immunzell-Aktivatoren zusammenlagern und dann synergistisch wirken. Es kann als Adjuvans dienen, also Immunreaktionen unspezifisch verstärken. Auch an der Wundheilung im Anschluss an eine Immunreaktion wirkt es mit. Manche Forscher sehen in ihm den zentralen Koordinator oder Choreografen steriler und infektionsbedingter Entzündungen.
Polygamie
Extrazelluläres HMGB1 bindet an mindestens elf verschiedene Transmembran-Rezeptoren, von denen wir hier nur wenige besprechen, nämlich TLR4, RAGE und CD24/Siglec-10. Einige Rezeptoren auf Zellen der angeborenen Abwehr lösen, wenn HMGB1 an ihre Außenseite bindet, innerzelluläre Signalketten wie den NFκ-B-Weg aus, die zur Produktion von entzündungsfördernden Zytokinen führen. Andere Rezeptoren senden dagegen nach HMGB1-Bindung produktionshemmende Signale ins Zellinnere. Darauf komme ich im Abschnitt „Hase und Schildkröte“ zurück.
Es lagert sich auch gerne mit anderen extrazellulären Molekülen zusammen, etwa mit DNA (natürlich!), aber auch mit dem Zytokin Interleukin-1, mit dem Chemokin CXCL12 oder bakteriellen Substanzen wie LPS. Bindet es dann gemeinsam mit ihnen an einen Rezeptor auf einer Immunzelle, so fällt die Wirkung oft stärker oder anders aus als bei einer Bindung des einen oder anderen Partners allein.
So lagert es sich bei einer Bakterieninfektion mit dem bakteriellen Molekül LPS zusammen. Durch das gemeinsame Andocken an einen Rezeptor einer antigenpräsentierenden Zelle wird ein Transportmechanismus ausgelöst, durch den das LPS ins Innere der Zelle gelangt, wo es zu präsentationsfähigen Antigen-Peptiden weiterverarbeitet wird, die dann spezifisch passende T-Zellen aktivieren, sodass die erworbene Abwehr ins Spiel kommt.
Der Rezeptor RAGE ist ebenso polygam wie sein Bindungspartner HMGB1: Zum einen bindet er auch eine Reihe weiterer Moleküle, zum anderen hängt seine Wirkung vom Zusammenspiel mit weiteren Rezeptoren auf denselben Zellen ab. Ein Regulierungsnetzwerk aus HMGB1, seinen extrazellulären Bindungspartnern und seinen Rezeptoren wird rasch unüberschaubar komplex. Konzentrieren wir uns auf drei exemplarische HMGB1-Regulierungsmechanismen: die Freisetzung, den extrazellulären Oxidations-Countdown und das innerzelluläre Wettrennen von Hase und Schildkröte.
Der schlechte und der gute Tod: Nekrose und Apoptose
Um die Logik des ersten Regulierungsmechanismus zu verstehen, benötigen wir Basiswissen über den traumatischen und den programmierten Zelltod, Nekrose und Apoptose genannt. Tatsächlich können Zellen noch auf andere Weisen sterben, etwa durch Pyroptose, ein für innerzelluläre Infektionen typisches Todesprogramm, oder durch NETose, eine Art Selbstmordattentat von Neutrophilen und anderen Granulozyten. Hier beschränke ich mich auf die beiden klassischen Formen des Ablebens. Bei der Nekrose, die unvorhergesehen kommt, schlägt die Zelle leck:

Dabei treten Moleküle wie DNA und allerlei Proteine aus, die normalerweise außerhalb von Zellen nichts zu suchen haben und daher prädestiniert sind, das Immunsystem als DAMP über den Gewebeschaden zu informieren. (Leider können auch T-Zellen und Antikörper auf diese Substanzen überreagieren, wenn sie sie fälschlich als körperfremd auffassen. Das war hier im Blog schon öfter Thema.)
Bei der Apoptose leitet eine Zelle selbst ihren kontrollierten Rückbau und ihre Entsorgung ein, etwa weil sie merkt, dass sie von Viren oder Bakterien befallen, alt oder auf andere Weise geschädigt ist. Sie ruft dazu eine T-Zelle zur Hilfe, die das Rückbauprogramm startet, und ihre Überreste werden von Makrophagen vertilgt:

Bei der Apoptose sollten keine Zellinnereien auslaufen. In der Spätphase kann es aber zu einer sogenannten sekundären Nekrose kommen, bei der das in kleinem Umfang doch passiert. Außerdem scheiden apoptotische Zellen aktiv Substanzen aus, etwa um das Immunsystem über die Ursache ihres Ablebens zu informieren oder um Makrophagen – ihre Totengräber – anzulocken.
Freisetzung von HMGB1: passiv, schnell und reduziert – oder aktiv, verzögert und oxidiert
Bei einer Nekrose tritt HMGB1 passiv aus einer sterbenden Zelle aus, sobald diese undicht wird. Viele nekrotische Zellen haben keine Zeit, das Molekül vorher zu modifizieren. Dann gelangt das HMGB1 in genau der Form in die Gewebsflüssigkeit, in der es im Zellinneren vorlag.
Stirbt eine Zelle dagegen durch Apoptose oder Pyroptose, kann sie das HMGB1 in Ruhe umbauen, um es auf seine künftige Aufgabe vorzubereiten. Zu diesen sogenannten posttranslationalen Modifikationen des Proteins zählen etwa die oben erwähnte Acetylierung oder Phosphorylierung, die für einen geregelten Transport aus dem Zellkern ins Zytoplasma und aus dem Zytoplasma in den extrazellulären Raum sorgen.
Jetzt kommen die drei Thiol-Reste der Cysteine im HMGB1-Molekül ins Spiel, die ich oben im Steckbrief erwähnt habe. Sollte das Schulwissen über Redox-Zustände verschüttet sein, hier eine kurze Auffrischung: Oxidation wurde klassisch als Hinzufügen von Sauerstoff (O wie Oxygenium) oder als Entzug von Wasserstoff (H wie Hydrogenium), also als Dehydrierung aufgefasst. Der umgekehrte Vorgang, also die Hinzufügung von Wasserstoff oder der Entzug von Sauerstoff, wird Reduktion genannt.
Sinnvoller ist es eigentlich, diese sogenannten Redox-Reaktionen als Abgabe oder Aufnahme von Elektronen zu definieren, aber wir bleiben hier beim volkstümlichen Verständnis: Ein frisch aufgeschnittener Apfel hat helles Fruchtfleisch. An der Luft, also bei der Reaktion mit Sauerstoff, oxidiert er. Die Schnittfläche läuft an und wird schließlich richtig braun:

Antioxidantien wie Vitamin C können diesen Prozess aufhalten; sie sind Reduktionsmittel.
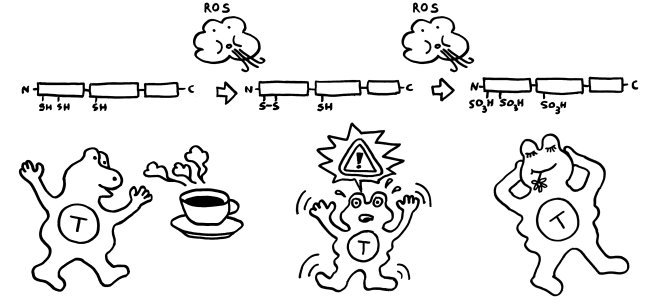
Im Normalfall ist das chemische Milieu in einer Zelle reduzierend; es gibt kaum freien Sauerstoff, der HMGB1 oxidieren könnte. Daher liegen im Zellkern alle drei Cystein-Reste als Thiol (-SH) vor. Bei der abrupten Freisetzung aus einer nekrotischen Zelle bleibt das Molekül zunächst in dieser vollständig reduzierten Form.
Gerät das Molekül jedoch in ein oxidierendes Milieu, so verändert es sich schrittweise: Zunächst bildet sich zwischen den beiden benachbarten Resten in Box A eine Disulfidbrücke (-S-S-) aus; die Reste geben also Wasserstoff ab; der dritte Rest liegt weiter als Thiol (-SH) vor. Wird das Molekül weiterhin mit reaktionsfreudigem Sauerstoff konfrontiert, reagieren schließlich alle drei Reste mit ihm und bilden Sulfonsäure (-SO3H).
Diese schrittweise Oxidation kann sogar innerhalb der Zelle ablaufen, wenn sich das chemische Milieu ändert: Manchmal kann das HMGB1 aus nekrotischen Zellen eine Disulfidbrücke ausbilden, bevor es freigesetzt wird. Auch Makrophagen oder dendritischen Zellen, die mit Bakterien infiziert sind und einen programmierten Zelltod namens Pyroptose durchlaufen, enthalten reaktionsfreudigen Sauerstoff und setzen HMGB1 mit einer Disulfidbrücke frei. Bei einer Apoptose wird HMGB1 vor seiner aktiven Ausscheidung oft sogar vollständig oxidiert, denn die Mitochondrien in den sterbenden Zellen setzen bei ihrem Rückbau reaktiven Sauerstoff frei.
Die drei freigesetzten Formen von HMGB1 wirken sich auf das Immunsystem grundverschieden aus, wie ich im nächsten Abschnitt zeige.
Countdown durch Oxidation
Vollständig reduziertes HMGB1 (das mit den drei Thiol-Resten), wie es etwa bei einer Nekrose freigesetzt wird, wirkt als Lockstoff, und zwar in Verbindung mit einem anderen Molekül: Es bindet an das Chemokin CXCL12, das aus aktivierten Immunzellen abgeschieden wird. Das Doppelpack (ein sogenanntes Heterodimer) aus HMGB1 und CXCL12 bindet sehr gut an den Chemotaxis-Rezeptor CXCR4 und führt dazu, dass Immunzellen mit diesem Rezeptor angelockt werden und sich vor Ort vermehren. Findet das reduzierte HMGB1 nicht genug CXCL12-Moleküle vor, kann es durch Bindung an den Rezeptor RAGE sogar die CXCL12-Produktion ankurbeln. Diese Rekrutierung von Immunzellen ist nach einer Nekrose besonders sinnvoll, denn diese Form des Zelltods läuft – wie oben zu sehen war – ungeplant und ohne Beteiligung von Immunzellen ab. Und die Überreste der zugrunde gegangenen Zellen müssen ja beseitigt, die Ursachen bekämpft, die Gewebeschäden repariert werden.
Teilweise oxidiertes HMGB1, das vorne eine Disulfidbrücke und hinten eine Thiolgruppe aufweist, kann dagegen an den Immunzell-Rezeptor TLR4 binden und so die Produktion und Freisetzung von Zytokinen anstoßen, also eine Entzündungsreaktion anheizen. Das ist besonders sinnvoll, wenn bereits Immunzellen vor Ort sind, von denen einige gerade an einer Pyroptose zugrunde gehen: Hier müssen keine weiteren Immunzellen rekrutiert, sondern die anwesenden zur eifrigen Mitarbeit an der Entzündung angeregt werden.
Vollständig oxidiertes HMGB1 hingegen, bei dem alle drei Cystein-Reste als Sulfonsäure vorliegen, kann Immunzellen weder anlocken und zur Vermehrung anregen noch zur Zytokinproduktion bewegen. Es bindet weder an CXCL12 und CXCR4 noch an TLR4: Es trägt überhaupt nichts zu einer Entzündungsreaktion bei, sondern stimmt anwesende Immunzellen tolerant, sozusagen als Anti-DAMP. Diese Wirkung ist besonders sinnvoll, wenn eine Zelle eine geregelte Apoptose durchlaufen hat, denn hier ist keine weitere Immunreaktion nötig: Es reicht, wenn die bereits anwesenden Fresszellen den Rest der gestorbenen Zelle beseitigen.
Zumeist wird HMGB1 also genau in der Form freigesetzt, in der es die jeweils erforderlichen Reaktionen in Gang setzen kann. Analog zum oxidierenden Apfel von oben sind hier die drei Formen des Moleküls dargestellt, ergänzt um die beiden Oxidationsreaktionen durch reaktiven Sauerstoff (ROS) und um die jeweilige Hauptwirkung des Moleküls auf das Immunsystem, hier durch eine T-Zelle vertreten: Anlockung, Aktivierung und Toleranz:
Wie verhindert unser Körper aber, dass das vollständig reduzierte HMGB1, das aus nekrotischen Zellen austritt, immer mehr und mehr Immunzellen rekrutiert und alarmiert, sodass die Entzündung chronisch wird und das umliegende Gewebe Schaden nimmt? Nun: Das extrazelluläre chemische Milieu ist im Vergleich zum gesunden Zellinneren oxidativ. Mit der Freisetzung von vollständig reduziertem HMGB1 beginnt daher ein Countdown. Die extrazelluläre Flüssigkeit oxidiert die Moleküle zunächst teilweise, dann vollständig – bis sie nicht mehr als Alarmsignale wirken.
Hase und Schildkröte
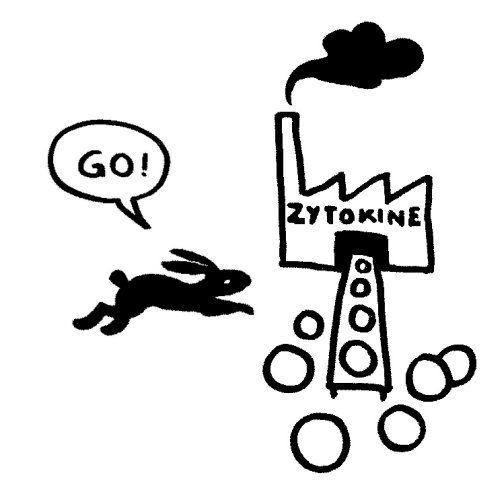
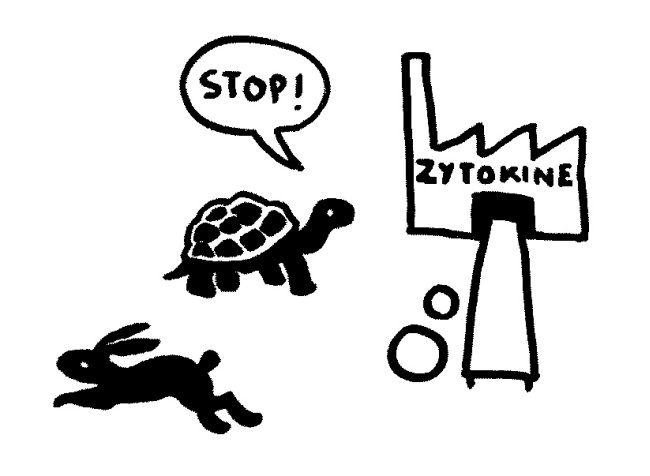
Wir kommen zum dritten Kontrollmechanismus, nämlich der Beschränkung der Zytokinproduktion in Makrophagen und anderen Immunzellen, die HMGB1 in seiner teilweise oxidierten Form aktiviert. Es bindet nicht nur an aktivierende Rezeptoren wie TLR4, sondern auch an inhibierende Rezeptoren wie CD24 und Siglec-10. TLR4 löst daraufhin eine schnelle Signalkette aus, CD24/Siglec-10 eine langsamere:

Beide Signale streben den Chromosomen im Zellkern zu, um dort die Ablesung der Zytokin-Gene zu beeinflussen:

Das vom TLR4 ausgesandte Signal erreicht sein Ziel zuerst und stößt eine starke Produktion entzündungsfördernder Zytokine an:
Etwas später trifft das Stoppsignal ein und beendet die Ablesung der Zytokin-Gene. Die Zelle lässt die Produktion auslaufen:
Bei Autoimmunerkrankungen versagen die checks and balances
So raffiniert diese Kontrollmechanismen sind: Bei Autoimmunerkrankungen versagen sie. Welchen Anteil hat HMGB1 an diesen Störungen? Es ist sicher nicht die Hauptursache, aber es kann in Individuen mit entsprechender Veranlagung eine Überaktivierung des Immunsystems fördern. Wichtiger als seine direkte Wirkung auf die Zellen der erworbenen Abwehr, also auf die Vermehrung und Aktivierung von T- und B-Zellen, ist vermutlich die indirekte, nämlich über die antigenpräsentierenden Zellen vermittelte Wirkung.
Sein Beitrag zum Teufelskreis sei am Beispiel Typ-1-Diabetes skizziert: Wenn Betazellen im Pankreas sterben, kann zunächst passiv ein wenig HMGB1 freigesetzt werden. Bei einer unzureichenden Entsorgung der Zellreste reifen durch dieses Alarmsignal oder DAMP zahlreiche dendritische Zellen und Makrophagen heran und werden aktiv: Zum einen können sie selbst noch mehr HMGB1 freisetzen und so die Entzündungsreaktion verstärken. Zu anderen aktivieren sie nun ihrerseits Lymphozyten, indem sie ihnen Antigene präsentieren – unter anderem jene Autoantigene, die zusammen mit dem HMGB1 aus den sterbenden Betazellen ausgetreten sind. In genetisch prädestinierten Individuen greifen diese autoreaktiven Lymphozyten daraufhin Betazellen an, was diese zum Absterben bringt, und so weiter und so fort.
Dieses Modell hat sich im Tierversuch bewährt: Die Pankreas-Lymphknoten von Mäusen mit Diabetes-Neigung, in denen HMGB1 experimentell neutralisiert wurde, enthielten weniger dendritische Zellen des an der Diabetes-Entstehung beteiligten Subtyps und dafür mehr regulatorische T-Zellen (Tregs). In der Milz der Tiere fanden sich zudem sogenannte tolerogene dendritische Zellen, die – wie die Tregs – besänftigend auf Lymphozyten einwirken und so den Teufelskreis durchbrechen können. Es gab zwar noch autoreaktive T-Zellen in den Pankreas-Lymphknoten, aber diese wurden mangels HMGB1 kaum noch in die Langerhans-Inseln gelockt und richteten daher unter den Betazellen wenig Schaden an.
Auch in Versuchstieren mit Lupus-Neigung beteiligt sich HMGB1 an der Pathogenese. Bei ihnen kommen vermutlich zwei Faktoren hinzu: Erstens richtet sich die Autoimmunreaktion bei Lupus primär gegen Zellkern-Bestandteile wie DNA oder Histone – und an diese bindet HMGB1 bekanntlich besonders gut. Es transportiert diese Autoantigene über den Rezeptor RAGE sehr effizient in die antigenpräsentierenden Zellen hinein, was die Autoantigenpräsentation und damit die Aktivierung autoreaktiver Lymphozyten verstärkt. Außerdem tragen auch die Endothelzellen unserer Blutgefäße den Rezeptor RAGE. Bindet dieser HMGB1, so können sich die Blutgefäße entzünden, und es kommt zu einer Lupus-Vaskulitis.
Bei rheumatoider Arthritis scheint zunächst ein Sauerstoffmangel (Hypoxie) Zellen in den Gelenken unter Stress zu setzen. Im Zuge ihrer Stressreaktion setzen sie HMGB1 in die Gelenkflüssigkeit frei, woraufhin sich die Gelenke entzünden können. Das Molekül fördert auch die Neubildung von Blutgefäßen und von Zellen, die Knochengewebe abbauen: sogenannten Osteoklasten. So kann sich die Gelenkschädigung verschlimmern.
Leider ist die Ausschaltung von HMGB1 kein Allheilmittel für Autoimmunerkrankungen. Zum einen hat das Molekül in unserem Körper zahlreiche weitere, lebenswichtige Funktionen. Zum anderen gibt es Hinweise, dass es bei chronischen Entzündungen nicht nur verheerend, sondern auch heilsam wirken kann. So scheint es bei einer Myositis – einer Entzündung von Muskelgewebe – anfangs die Entzündung zu fördern, später aber zum Schutz und zur Regeneration des Muskels beizutragen. Man müsste also eine HMGB1-Blockade entwickeln, die örtlich und zeitlich sehr begrenzt wirkt. Eingriffe in die komplexen und noch nicht vollständig verstandenen checks und balances des Immunsystems bleiben ausgesprochen heikel.
Literatur:
A. Broggi, F. Granucci (2015): Microbe- and danger-induced inflammation
H. E. Harris et al. (2012): HMGB1: A multifunctional alarmin driving autoimmune and inflammatory diseases
R. Kang et al. (2013): HMGB1 in Cancer: Good, Bad, or Both?
S.-A. Lee et al. (2014): The Role of High Mobility Group Box 1 in Innate Immunity
G. Li et al. (2013): HMGB1: the central cytokine for all lymphoid cells
M. Magna, D. S. Pisetsky (2014): The Role of HMGB1 in the Pathogenesis of Inflammatory and Autoimmune Diseases
C. Pilzweger, S. Holdenrieder (2015): Circulating HMGB1 and RAGE as Clinical Biomarkers in Malignant and Autoimmune Diseases
H. Yang et al. (2015): High Mobility Group Box Protein 1 (HMGB1): The Prototypical Endogenous Danger Molecule
J. Zhong (2012/2013): HMGB1 as an Innate Alarmin Promotes Autoimmune Progress: An Essential Role in the Pathogenesis of Type 1 Diabetes