Im letzten Beitrag habe ich ein Genregulierungsnetzwerk vorgestellt, das unter der Kontrolle des Transkriptionsfaktors VGLL3 steht. Es etabliert viele der Unterschiede zwischen männlichem und weiblichem Immunsystem, die zum hohen Frauenanteil unter den Patienten mit Autoimmunerkrankungen beitragen, aber auch zu höheren Krebs- und Infektionsrisiken von Männern.
Ein solches Netzwerk kann sich nach seiner Initialzündung rasch ausweiten, etwa wenn unter den Genen, deren Ablesung der erste Transkriptionsfaktor fördert, mindestens ein weiterer Transkriptionsfaktor ist, der wiederum die Expression zahlreicher Gene beeinflusst, und so weiter.
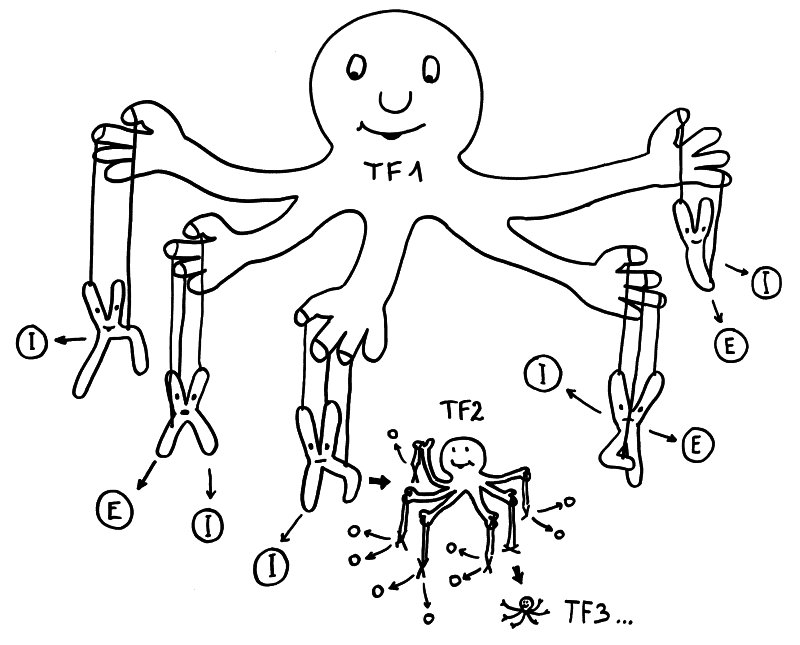
Geschlechtsspezifisches Regulierungsnetzwerk: Transkriptionsfaktor 1 fördert in weiblichen Zellen die Ablesung vieler Immunsystem- (I) und Entzündungsprozess-Gene (E) auf verschiedenen Chromosomen – und auch die Expression von Transkriptionsfaktor 2, der zahlreiche weitere Gene reguliert, usw. usf.
Eines allerdings blieb offen: Wie kommt der allererste Unterschied zustande, der wiederum für eine stärkere Expression von VGLL3 in weiblichen Zellen sorgt? Denn das Gen für diesen Transkriptionsfaktor sitzt nicht etwa auf dem X-Chromosom, sondern auf Chromosom 3, von dem sowohl weibliche als auch männliche Ungeborene zwei Exemplare haben.
Symmetriebrüche
Dieser Frage nach der Initialzündung gehe ich hier nach. Sie wird in der Fachliteratur auch unter der Bezeichnung „Symmetriebruch“ abgehandelt. Bis zu einem bestimmten Stadium verläuft die Entwicklung weiblicher und männlicher Embryonen gleich: Nachdem anfangs noch alle Zellen gleich aussehen und auch austauschbar sind, legen sich bestimmte Zellen auf ein „Schicksal“ fest und bilden bei ihren weiteren Teilungen Zellklone, aus denen die Anlagen eines Organs werden, etwa des Gehirns oder der Leber. Das ist der erste Symmetriebruch.
Schon bald danach entwickeln sich die Organanlagen je nach Geschlecht unterschiedlich weiter. Bei den Geschlechtsorganen ist das offenkundig, bei den meisten anderen Organen fällt der Unterschied viel subtiler aus. Das ist der zweite Symmetriebruch, der uns hier interessiert. Er betrifft auch die Bestandteile des Immunsystems, also etwa den Thymus oder die Vorläuferzellen der weißen Blutkörperchen – die sogenannten hämatopoetischen Stammzellen – in der embryonalen Leber und später im Knochenmark.
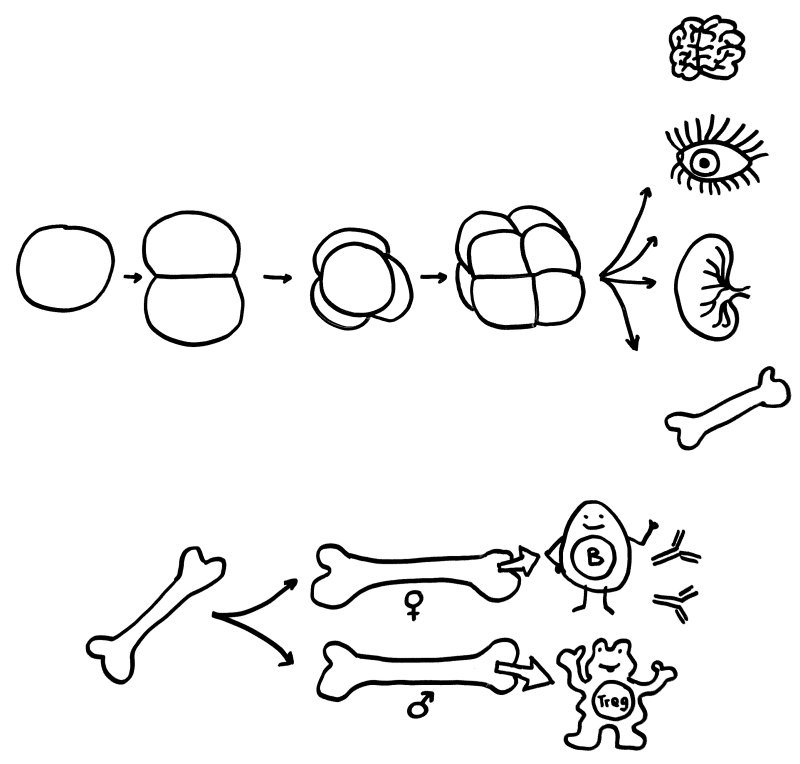
Oben: Der erste Symmetriebruch in der Embryonalentwicklung sorgt dafür, dass aus einer anfangs gleichförmigen Zellkugel letztlich unterschiedliche Organe werden. Unten: Ein zweiter Symmetriebruch lässt die Entwicklung der einzelnen Organe bei Frauen und Männern leicht unterschiedlich verlaufen – hier die Entwicklung des Knochenmarks und damit der Zellen unserer erworbenen Abwehr.
Diese subtil auseinanderlaufenden embryonalen Entwicklungswege führen später zu Geschlechtsunterschieden (sex bias) in der Grundausstattung des weiblichen und des männlichen Immunsystems, in den Immunreaktionen von Frauen und Männern auf verschiedene Antigene und schließlich in der Inzidenz und Prävalenz zahlreicher Autoimmunerkrankungen, die ich hier im Blog schon mehrfach behandelt habe – besonders ausführlich in dem Artikel Noch einmal: Geschlecht, Hormone, Immunsystem.
Weibliches und männliches Immunsystem
Geschlechtliche Immunsystem-Differenzen gibt es nicht nur bei Mensch und Maus, sondern auch etwa bei Seeigeln, Taufliegen oder Meisen. Damit ist bereits klar, dass sie sich nicht auf die erworbene Abwehr mit ihren Lymphozyten beschränken, die es ja nur bei Wirbeltieren gibt, sondern auch die angeborene Abwehr umfassen.
Beim Menschen treten solche Unterschiede in allen Lebensphasen zutage. Um zunächst die evolutionär ältere angeborene Abwehr zu betrachten: Im Mutterleib zeigen männliche Feten stärkere Entzündungsreaktionen als das weibliche Ungeborene. Auch in vorpubertären Jungen laufen Entzündungen noch heftiger ab als in Mädchen. Nach der Pubertät verkehrt sich dies, weil Testosteron in den meisten Situationen entzündungshemmend wirkt. Sowohl vorpubertäre Jungen als auch erwachsene Männer haben mehr natürliche Killerzellen (NK-Zellen) als Mädchen und Frauen; Senioren haben dagegen weniger NK-Zellen als Seniorinnen, deren angeborene Immunzellen außerdem mehr Interleukin-10 produzieren. Und dass auch die Menge und der Aktivierungsgrad der Mikrogliazellen im Nervensystem je nach Geschlecht und Lebensphase unterschiedlich ausfallen, habe ich kürzlich gegen Ende meines langen Artikels über den Schmerz ausgeführt.
In der evolutionär jüngeren erworbenen Abwehr, bei der Lymphozyten dank ihrer antigenspezifischen Aktivierung präziser gegen Krankheitserreger vorgehen können, sind die Unterschiede noch vielfältiger. So produzieren neugeborene Jungen mehr Antikörper des Typs IgE als neugeborene Mädchen. Im Blut vorpubertärer Jungen findet man auch mehr Antikörper des Typs IgA und IgM sowie mehr regulatorische T-Zellen (Tregs) als im Blut von Mädchen; andere Konzentrationen wie die der CD4+- und der CD8+-T-Zellen, der B-Zellen sowie der Antikörper des Typs IgG sind dagegen noch gleich. Nach der Pubertät gibt es in Frauen mehr CD4+- Zellen und weniger CD8+-Zellen als in Männern; T-Zellen lassen sich in ihnen leichter aktivieren und vermehren sich dann stärker; auch B-Zellen und ihre Produkte, die Antikörper, kommen in Frauen in größerer Zahl vor. Deshalb sprechen Frauen auch auf Impfungen etwas stärker an als Männer. Männer verfügen dafür weiterhin über mehr immunreaktionshemmende Tregs. All diese Unterschiede bleiben bis ins hohe Alter bestehen.
Krankheiten mit deutlichen sex bias
Unter den etwa 80 Autoimmunerkrankungen haben mehr als 20 einen starken und etwa 25 weitere einen moderaten Frauenüberhang. Deutlich mehr Frauen als Männer (bzw. Weibchen als Männchen) erkranken zum Beispiel an Morbus Basedow, Hashimoto-Thyreoiditis, Multipler Sklerose, rheumatoider Arthritis und systemischem Lupus erythematosus sowie an den Infektionserkrankungen, die durch HIV (AIDS), Influenza-Viren (Grippe), Zika-Viren, Legionellen oder Plasmodien (Malaria) ausgelöst werden.
Etwa 20 Autoimmunerkrankungen sind bei Männern häufiger als bei Frauen, etwa Morbus Bechterew. Die meisten dieser 20 Krankheiten sind aber so selten, dass in der Summe viel mehr Frauen von Autoimmunerkrankungen betroffen sind. Zu den Infektionen mit deutlichem Männerüberhang zählen Ebola, MERS, Hepatitis B, Tuberkulose, Leptospirose, Bilharziose oder Aspergilliose. Auch viele Krebserkrankungen sind bei Männern häufiger, etwa Leukämien oder Nieren-, Leber-, Lungen-, Speiseröhren-, Magen-, Darm- und Blasenkrebs. Krebserkrankungen der Geschlechtsorgane sind in dieser Geschlechterstatistik logischerweise ausgeklammert.
Eine Frage des Ressourceneinsatzes
Wahrscheinlich sind das deutlich höhere Risiko von Frauen, eine Autoimmunerkrankung zu bekommen, und das deutlich höhere Risiko von Männern, an bestimmten Krebs- oder Infektionserkrankungen zu sterben, der Preis für eine alles in allem evolutionär bewährte geschlechtsspezifische Optimierung von Ressourceneinsätzen. Über weite Strecken der Wirbeltier-, Säugetier- und schließlich Primaten-Evolution war Energie ein äußerst kostbares Gut.
Die Mütter sehr junger Kinder benötigen zum Beispiel ein besonders widerstandsfähiges Immunsystem – sonst raffen Infektionen nicht nur sie selbst, sondern in der Folge auch ihren Nachwuchs dahin. Im männlichen Organismus sind die mit der Nahrung aufgenommenen Kalorien unter Umständen besser in weitere Spermien und Paarungsakte investiert, zum Beispiel durch die Produktion von möglichst viel Testosteron – auch wenn das Immunreaktionen hemmt. Solche unterschiedlichen Life histories und trade-offs habe ich vor längerem schon einmal ausführlich erklärt.
Embryonen und Feten haben kein Gender
Zu einem gewissen Teil mögen Geschlechtsunterschiede in der Häufigkeit von Autoimmunerkrankungen auch auf Umweltfaktoren, etwa eine unterschiedliche Ernährung, Kosmetik oder berufliche Schadstoff-Exposition zurückgehen. Aber je früher eine Erkrankung ausbricht oder sich zumindest anbahnt, bei Autoimmunstörungen oftmals viele Jahre vor der Diagnose, desto unwahrscheinlicher ist ein Gender-Unterschied als Ursache. Insbesondere männliche und weibliche Embryonen, Feten und Neugeborene leben in sehr ähnlichen Umwelten. (Zur Erinnerung: Beim Menschen spricht man bis zur 9. Schwangerschaftswoche von Embryonen und dann – wenn alle inneren Organe angelegt sind – von Feten.) Daher kann man bei den frühesten Abweichungen zwischen männlichem und weiblichem Immunsystem getrost von einer Ursache im biologischen Geschlecht ausgehen.
Am Anfang steht die befruchtete Eizelle. Sie enthält fast nur Zytoplasma aus der mütterlichen Eizelle, denn der Spermienkopf ist zu klein, um viel beizusteuern. Auch der Chromosomensatz, den die Eizelle beisteuert, ist immer gleich: Er enthält 22 Autosomen (also Nicht-Geschlechtschromosomen) und ein X-Chromosom. Das Spermium bringt ebenfalls 22 Autosomen mit, deren Informationsgehalt vom Geschlecht des künftigen Kindes unabhängig ist. Die ersten Unterschiede zwischen weiblichen und männlichen Embryonen können also eigentlich nur vom Geschlechtschromosom herrühren, das das Spermium mitbringt: entweder ein zweites X oder ein Y.
Drei Möglichkeiten
Erstens: Das kleine Y-Chromosom enthält neben einigen Genen, die in ähnlicher Form auch auf dem X-Chromosom vorkommen, 25 „Männergene“, zu denen es im weiblichen Genom keine Entsprechungen gibt – darunter Sry, das Gen, das die Entwicklung männlicher Geschlechtsorgane anstößt. In den entstehenden Hoden läuft dann in der 10. Schwangerschaftswoche die Produktion des männlichen Sexualhormons Testosteron an, das die Entwicklung der Lymphorgane, des Gehirns und weiterer Organe beeinflusst. Auch weitere „Männergene“ auf dem Y-Chromosom könnten an einer etwas anderen Entwicklung männlicher Embryonen und ihres Immunsystems beteiligt sein.
Zweitens: Einige Gene auf dem Y-Chromosom haben sich im Lauf der Evolution (zum Teil mehrfach) verdoppelt und kommen heute in variabler Anzahl vor. Die Kopienzahl dieser sogenannten Y-chromosomalen Multicopy-Gene beeinflusst zum einen die Funktionsfähigkeit von Spermien, die ein Y-Chromosom enthalten, bzw. die Lebensfähigkeit der mit ihnen gezeugten männlichen Embryonen und Feten, und zum anderen das Risiko der Töchter, bestimmte Autoimmunerkrankungen zu bekommen – obwohl diese Töchter ja gar kein Y-Chromosom vom Vater erben. So haben Frauen, die MS, SLE oder rheumatoide Arthritis haben, deutlich seltener männliche Geschwister als gesunde Frauen, aber auch als Frauen mit Autoimmunerkrankungen, die bei beiden Geschlechtern gleich häufig auftreten. Diesem höchst seltsamen und noch nicht erschöpfend erforschten Phänomen, das als paternaler Parent-of-origin-Effekt bezeichnet wird, widme ich den nächsten Beitrag. Vielleicht verändert die Y-chromosomale Genkopienzahl in den Spermien-Vorläuferzellen des Vaters die epigenetische Markierungen und damit die Ablesbarheit der Gene des X-Chromosoms, das er zum Genom seiner Töchter beiträgt. Das könnte das Immunsystem der Töchter prägen und ihm im Extremfall von Anfang an eine gefährliche Schlagseite geben.
Eine dritte Möglichkeit: Zwar werden am 16. Tag der Schwangerschaft in allen Zellkernen eines weiblichen Embryos weite Teile eines der beiden X-Chromosomen stillgelegt, damit die darauf liegenden Gene im weiblichen Embryo oder Fetus nicht doppelt so stark abgelesen werden wie im männlichen. Aber beim Menschen entgehen etwa 15 Prozent der Gene auf dem zweiten X-Chromosom dieser Inaktivierung, sodass auch die höhere Dosis der Produkte dieser Gene zur weiblichen Entwicklung des Immunsystems beitragen könnte. Die Hypothese, dass eine unvollständige oder instabile X-Chromosom-Inaktivierung Autoimmunerkrankungen bei Frauen begünstigen könnte, habe ich hier schon einmal vorgestellt.
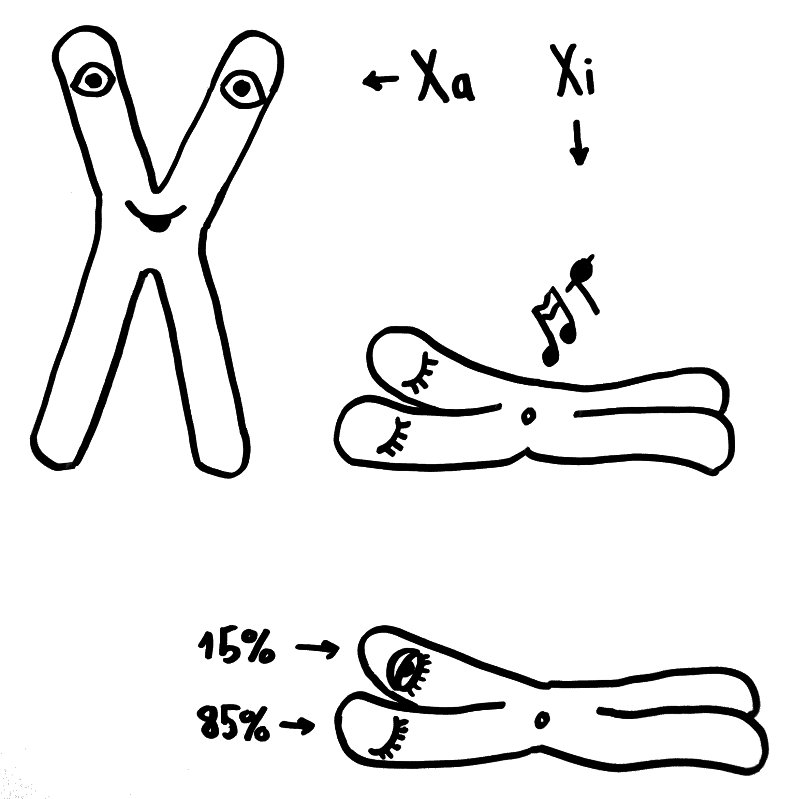
Oben: Von den beiden X-Chromosomen in den Zellen eines weiblichen Embryos bleibt eines aktiv (Xa), das andere wird inaktiviert (Xi). Unten: 15 Prozent der Gene auf dem Xi bleiben aber ablesbar.
Die Vermittler: Transkriptionsfaktoren, DNA-Methylierung, Micro-RNA und Sexualhormone
Ob die allerersten das Immunsystem betreffenden Unterschiede nun vom X- oder vom Y-Chromosom ausgehen: Irgendwie müssen sie ein Schneeballsystem in Gang setzen, denn auf fast allen Chromosomen des Menschen sind Gene zu finden, die bei den Geschlechtern unterschiedlich stark abgelesen werden und etwas mit dem Immunsystem zu tun haben. Wie eingangs festgestellt, kann der Transkriptionsfaktor VGLL3 diese Kaskade nicht anstoßen, denn sein Gen sitzt auf Chromosom 3, nicht auf einem Geschlechtschromosom. Aber vielleicht gibt es auch X- oder Y-chromosomale Genprodukte, die als Transkriptionsfaktoren fungieren und ein geschlechtsspezifisches Genregulierungsnetzwerk in Gang setzen.
Zur Etablierung der ersten geschlechtlichen Unterschiede im Immunsystem können neben solchen Transkriptionsfaktoren drei weitere Mechanismen beitragen:
- Epigenetische Markierungen wie Methylgruppen an der DNA beeinflussen die Ablesbarkeit von Genen. Die Gesamtheit aller Methylierungen eines Genoms, das sogenannte Methylom, weist schon bei der Geburt eines Menschen geschlechtsspezifische Unterschiede auf.
- Geschlechtshormone aktivieren Transkriptionsfaktoren und steuern so ebenfalls die Ablesestärke von Genen. Da Testosteron nicht etwa erst in der Pubertät produziert wird, sondern bereits vor der Geburt eines Jungen und in seiner ersten Lebensphase, kann dieses Hormon durchaus an der Ausbildung des frühen sex bias im Immunsystem mitwirken.
- Kurze RNA-Stränge, sogenannte Micro-RNAs oder miRNAs, binden an die Transkriptionsprodukte von Genen – die Messenger-RNA – und blockieren so die Proteinherstellung – auch die Herstellung von Immunsystem-Komponenten. Die Produktion der miRNAs wiederum wird von den Geschlechtschromosomen und -hormonen gesteuert.
Diese drei Mechanismen schließen einander also nicht aus, sondern greifen ineinander. In den folgenden Beiträgen stelle ich sie genauer vor.
Literatur
L. K. Case et al. (2015): Copy Number Variation in Y Chromosome Multicopy Genes Is Linked to a Paternal Parent-Of-Origin Effect on CNS Autoimmune Disease in Female Offspring
R. C. Chiaroni-Clarke et al. (2016): Sex bias in paediatric autoimmune diseases – Not just about sex hormones? (Bezahlschranke)
R. Dai, S. A. Ahmed (2014): Sexual dimorphism of miRNA expression: a new perspective in understanding the sex bias of autoimmune diseases
S. L. Klein, K. L. Flanagan (2016): Sex differences in immune responses
H. Lee et al. (2014): Reprogramming the Methylome: Reprogramming the Methylome: Erasing Memory and Creating Diversity
M. Suderman et al. (2017): Sex-associated autosomal DNA methylation differences are wide-spread and stable throughout childhood
J. Wang et al. (2016): Unusual maintenance of X chromosome inactivation predisposes female lymphocytes for increased expression from the inactive X