



1. Eine antigenpräsentierende Zelle (hier eine dendritische Zelle) gewinnt ein Antigen aus einem Pathogen. Die Infektion bemerken wir oft gar nicht; sie ist »stumm« oder »maskiert«.
2. Die antigenpräsentierende Zelle zeigt das Antigen und einen Kostimulator (die Kerze) vor. T-Helferzellen mit passendem T-Zell-Rezeptor werden aktiviert.
3. Die T-Helferzellen aktivieren B-Zellen mit derselben Antigen-Spezifität.
4. Die B-Zellen stellen Antikörper gegen das Antigen her und bekämpfen so die Infektion.
5. Einige T-Zellen überwinden die Blut-Hirn-Schranke und verwechseln Teile der Myelinscheiden um die Nervenzellen mit dem Pathogen-Antigen.
6. Myelinscheiden sind fettreiche Membranen von Schwann-Zellen: Gliazellen, die um Axone (Nervenzellausläufer) gewickelt sind und eine Isolationsschicht bilden. Sie sind für die
Weiterleitung von Nervenimpulsen notwendig. Links ein Längsschnitt durch ein Axon und seine Myelinscheide, rechts ein Querschnitt.
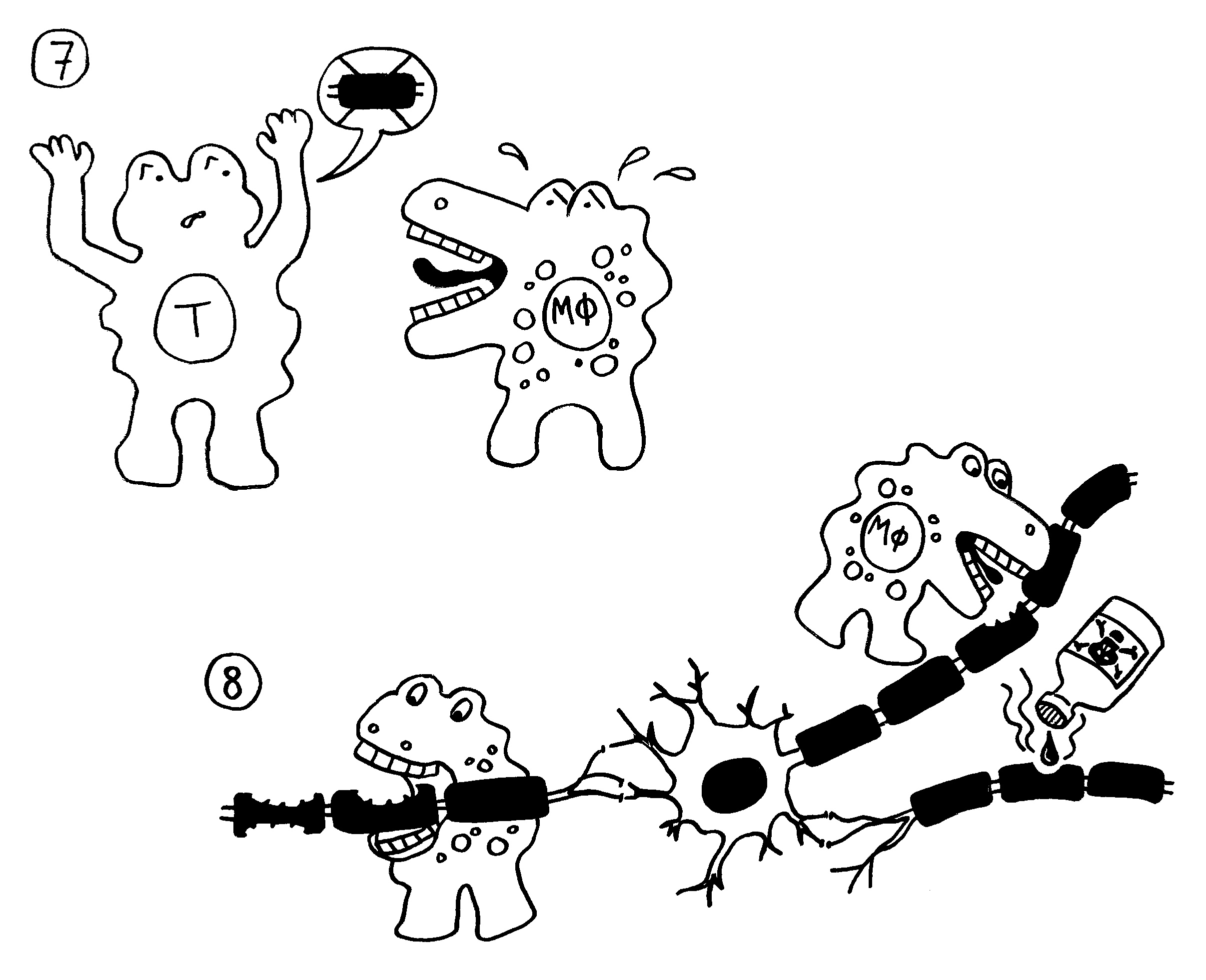
7. Die autoreaktiven T-Zellen rekrutieren Zellen der angeborenen Abwehr, zum Beispiel Makrophagen.
8. Die angelockten Immunzellen greifen die Myelinscheiden an. Das kann zu einer Lähmung
führen.
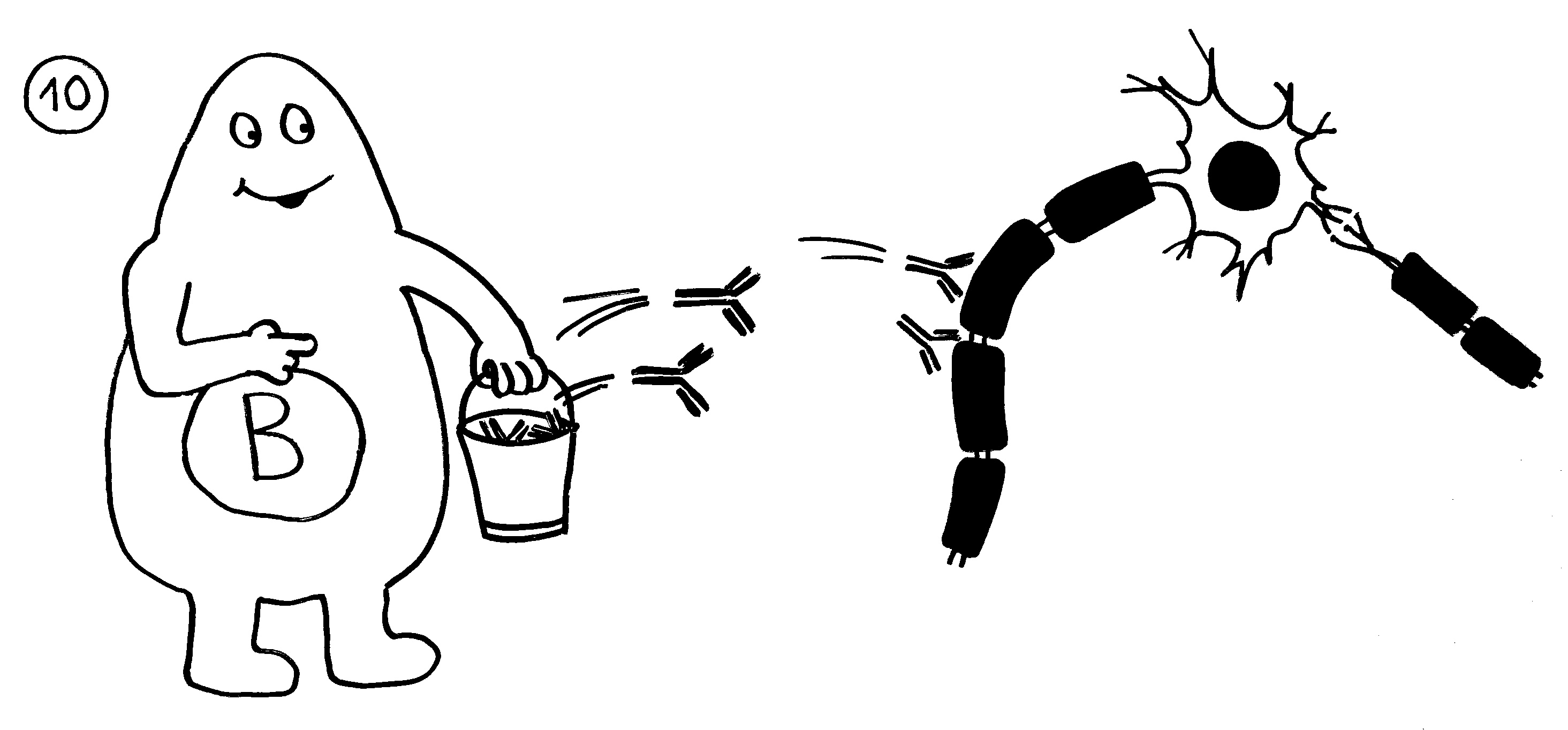
9. Bei einigen Immunneuropathien aktivieren autoreaktive T-Helferzellen auch autoreaktive
B-Zellen.
10. Die B-Zellen stellen Autoantikörper her, die an Myelinscheiden binden und so die Attacken anderer Immunzellen verstärken. – Medikamente oder die Selbstregulation des Immunsystems können die Angriffe rechtzeitig beendet. Dann bauen überlebende Gliazellen die Myelinscheiden allmählich wieder auf. Die Nerven können wieder Impulse weiterleiten; die Lähmung geht zurück.
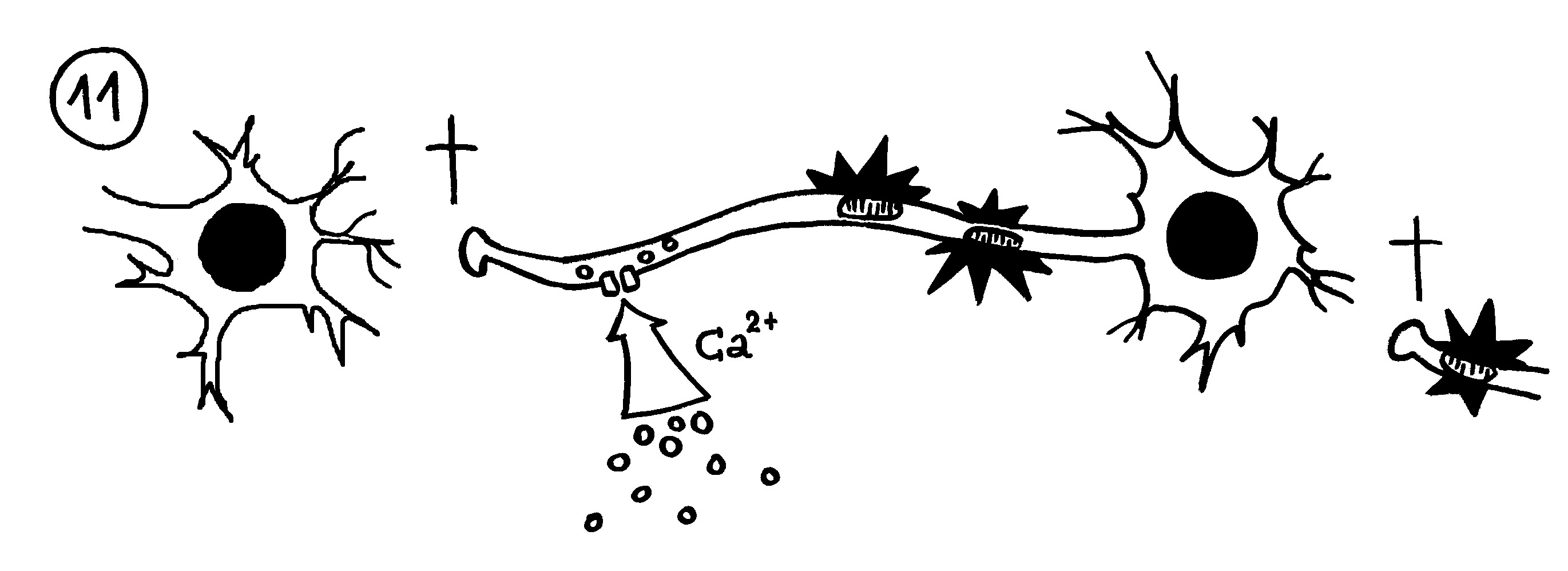
11. Bleibt die Myelinscheide dagegen defekt, strömen durch Ionenkanäle massenhaft Ionen (z. B. Kalzium) in die Nervenzellen ein. Die Mitochondrien schwellen an und schädigen die Axone (Sterne). Dann sterben die Axon-Enden (Kreuze), und der Kontakt zu anderen Nervenzellen bricht ab.
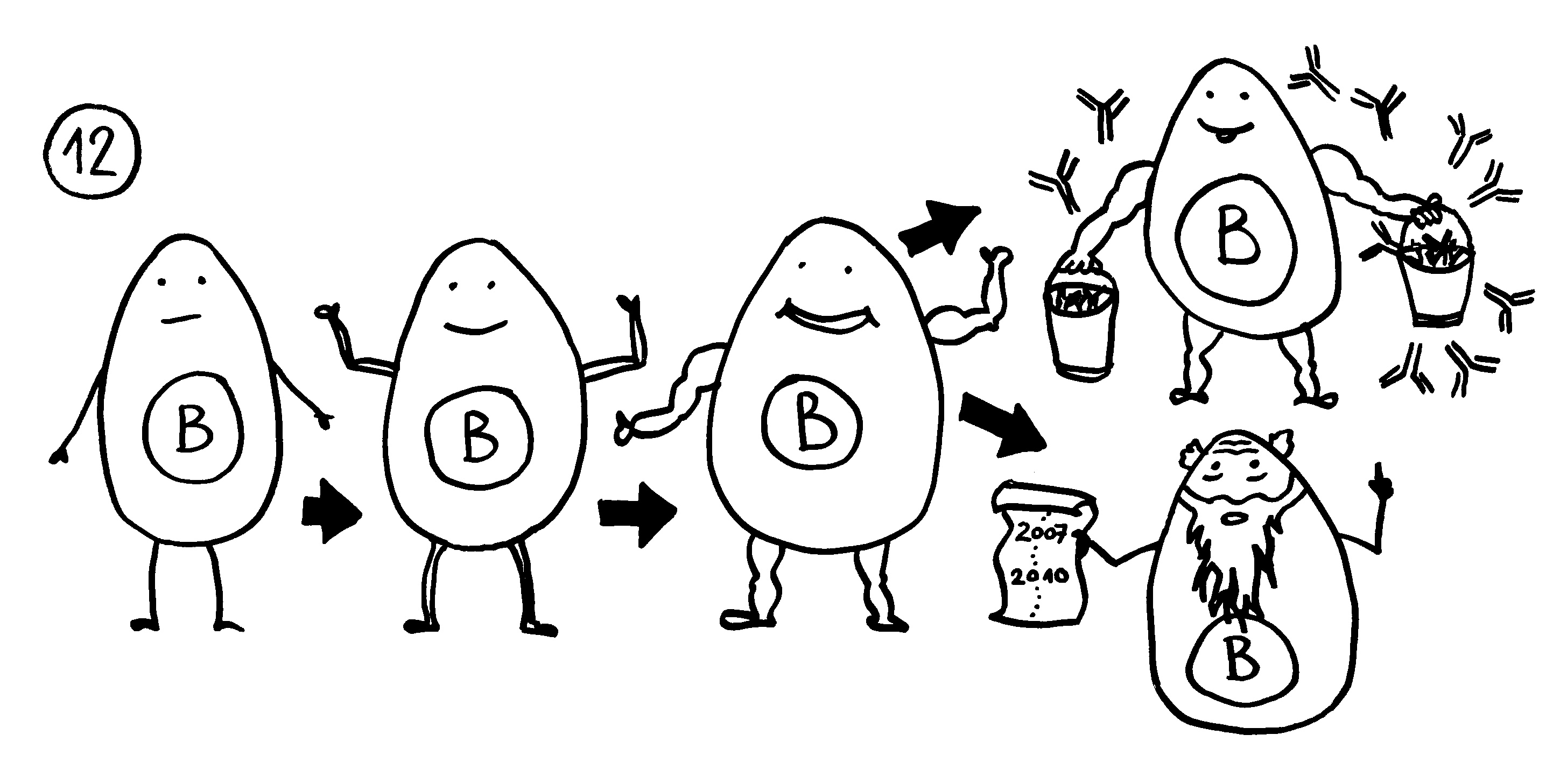
12. In der Nähe können sich Lymphfollikel bilden, in denen autoreaktive B-Zellen eine Affinitätsreifung durchlaufen. Außer antikörperproduzierenden Plasmazellen entstehen dabei Gedächtniszellen, durch die die Autoimmunreaktion chronisch werden kann.
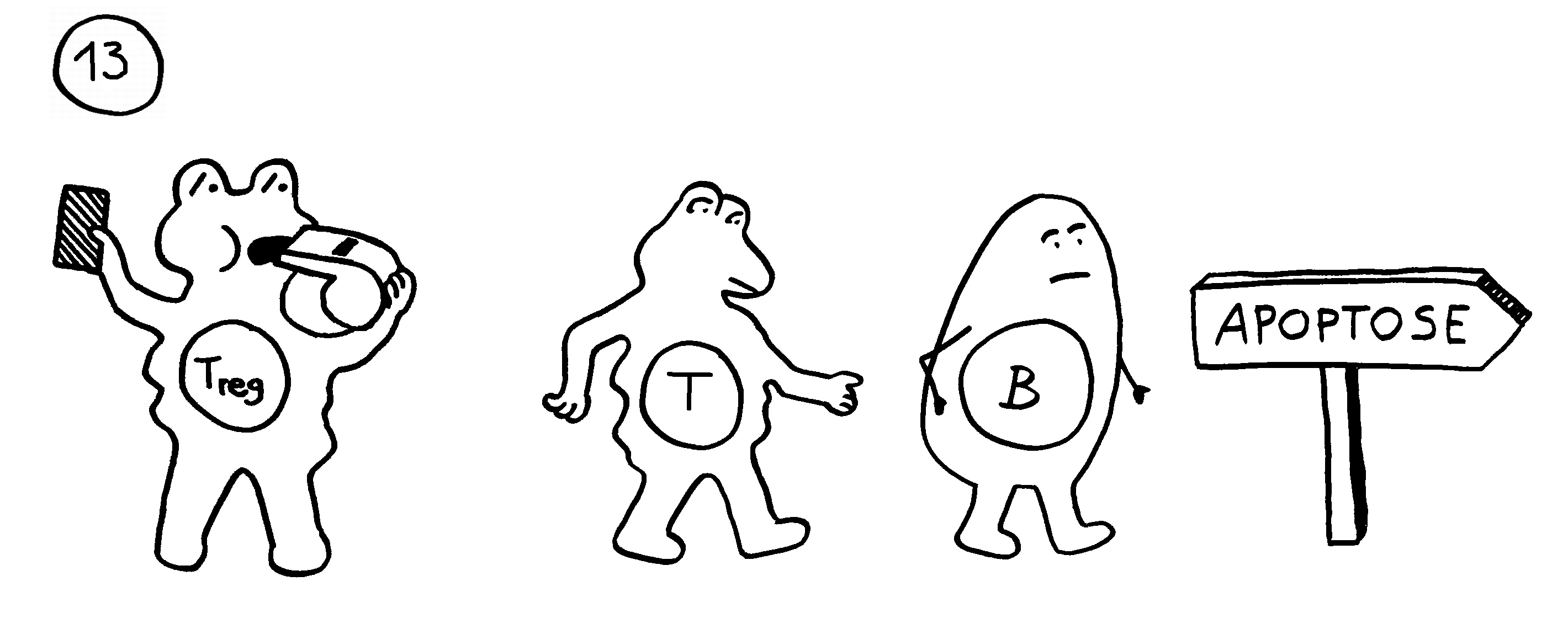
13. In anderen Fällen verhindern regulatorische T-Zellen die Chronifizierung: Sie schicken die autoreaktiven Lymphozyten rechtzeitig vom Platz und beenden die Immunreaktion.
Sie dürfen diese Zeichnung gerne in Folien etc. übernehmen, sofern Sie die Quelle angeben: Dr. Andrea Kamphuis, https://autoimmunbuch.de




