 #NaNoWriMo22, Tag 5
#NaNoWriMo22, Tag 5
Die Tiefsee – unendliche Weiten. Wir befinden uns in einer fernen Vergangenheit. Dies sind die Abenteuer eines Tiefseeangler-Weibchens, das viele Meilen von der Wasseroberfläche entfernt unterwegs ist, um fremde Männchen zu entdecken …
Im Ernst: Wie findet man da unten in der finsteren, kalten, erdrückenden Leere Partner? Die Tiefseeanglerfische (Ceratioidei) mussten sich nicht nur an den enormen Druck der auf ihnen lastenden Wassersäule anpassen, der normale Proteine in kürzester Zeit mit Wassermolekülen durchsetzen, verformen und funktionsuntüchtig machen würde, an die völlige Dunkelheit ihres Lebensraums und an den Mangel an Nahrung, sondern auch Mittel und Wege finden, um die Wahrscheinlichkeit von Begegnungen mit Artgenossen des anderen Geschlechts zu erhöhen.
Die Weibchen tragen am Ende eines angelförmigen Auswuchses an der Stirn Laternen, in denen symbiotische Leuchtbakterien etwas Licht für sie produzieren. Damit locken sie nicht nur Krebse und Fische an, die sie dann mit ihren riesigen Mäulern verschlingen, sondern senden auch den Männchen ein Zeichen. Das klappt aber nur, wenn diese schon in der Nähe sind.
Die Männchen sind winzig klein und haben, solange sie Junggesellen sind, große Augen und einen sehr empfindlichen Geruchssinn: Sie können noch kleinste Mengen der Sexuallockstoffe erschnuppern, die die Weibchen absondern, und stöbern ihre künftige Partnerin so auf. Den letzten Meter bewältigen sie dann mithilfe ihrer Augen und des Laternen-Leuchtfeuers. Bei vielen Tiefseeanglerfisch-Arten docken sie dann mit dem Mund an das Weibchen an, um es nicht wieder zu verlieren: manche nur vorübergehend, andere permanent; sie verwachsen regelrecht mit ihren und werden von da an über den Blutkreislauf des Weibchens mit Nährstoffen versorgt – bis dass der Tod sie scheidet. Ihre eigenen Organe verkümmern, bis auf die Hoden. Es gibt sogar einige Arten, bei denen mehrere Männchen mit einem Weibchen verwachsen; bis zu acht hat man schon entdeckt. So steht immer Sperma zur Verfügung, wenn das Weibchen ablaicht.
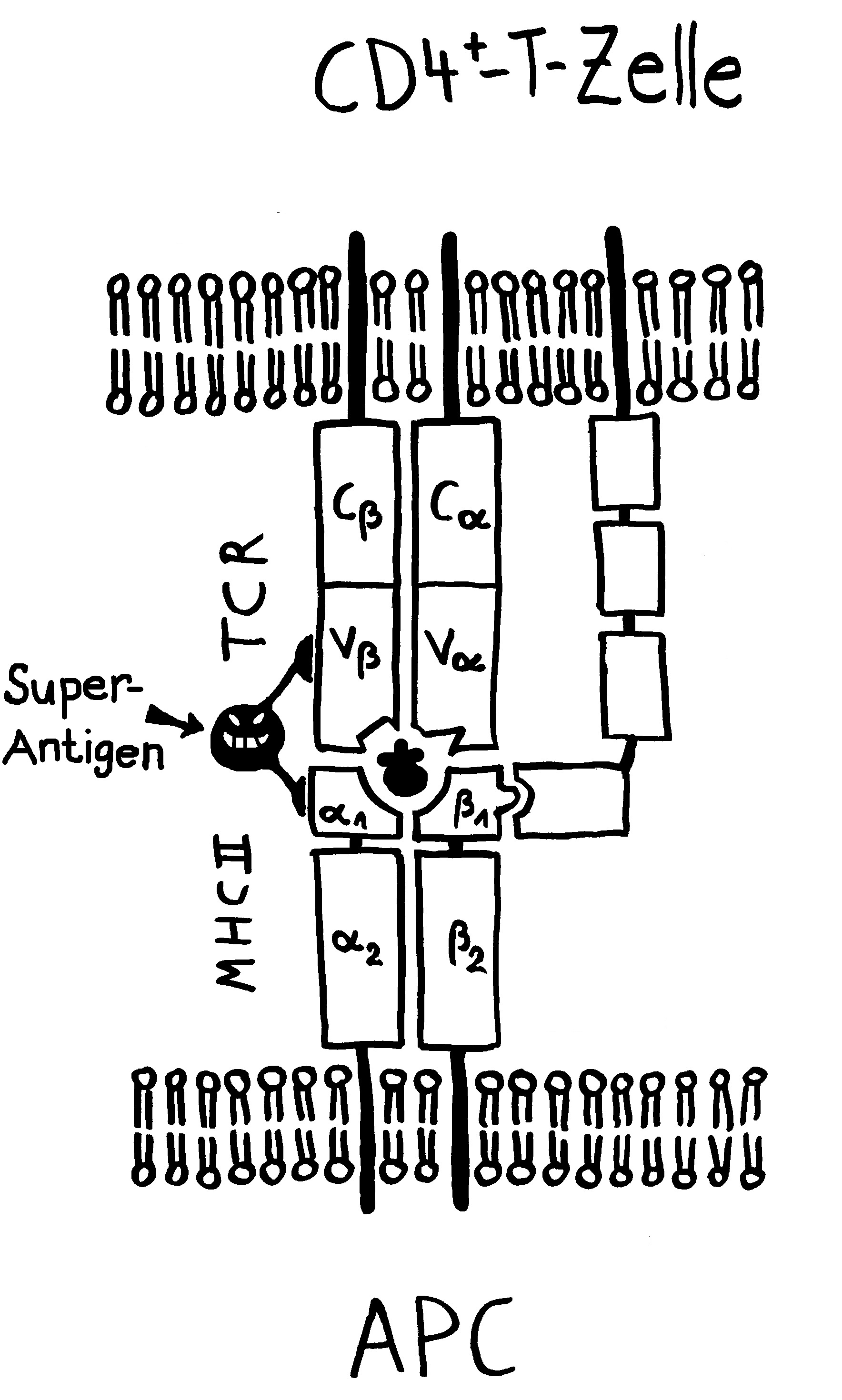
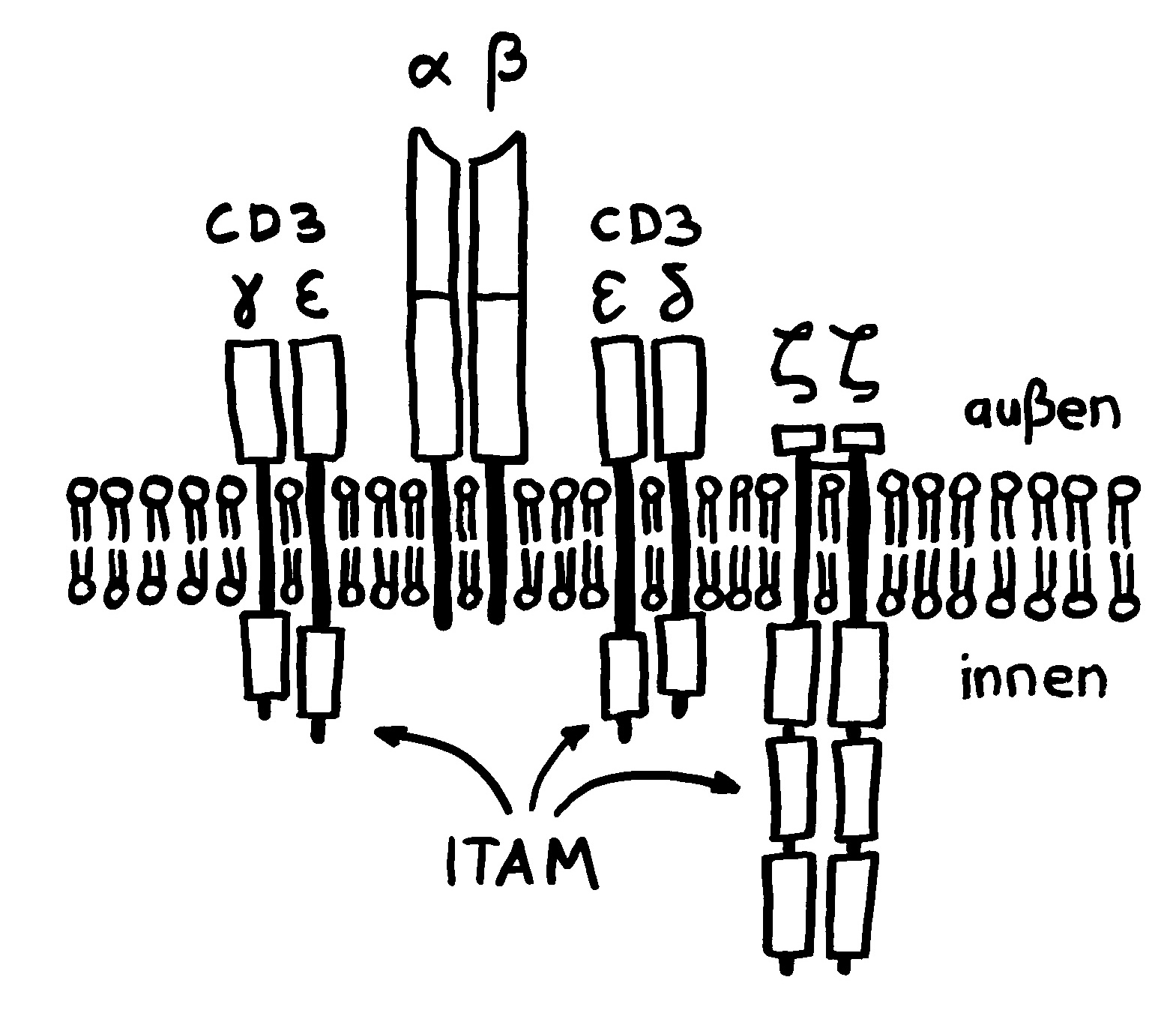
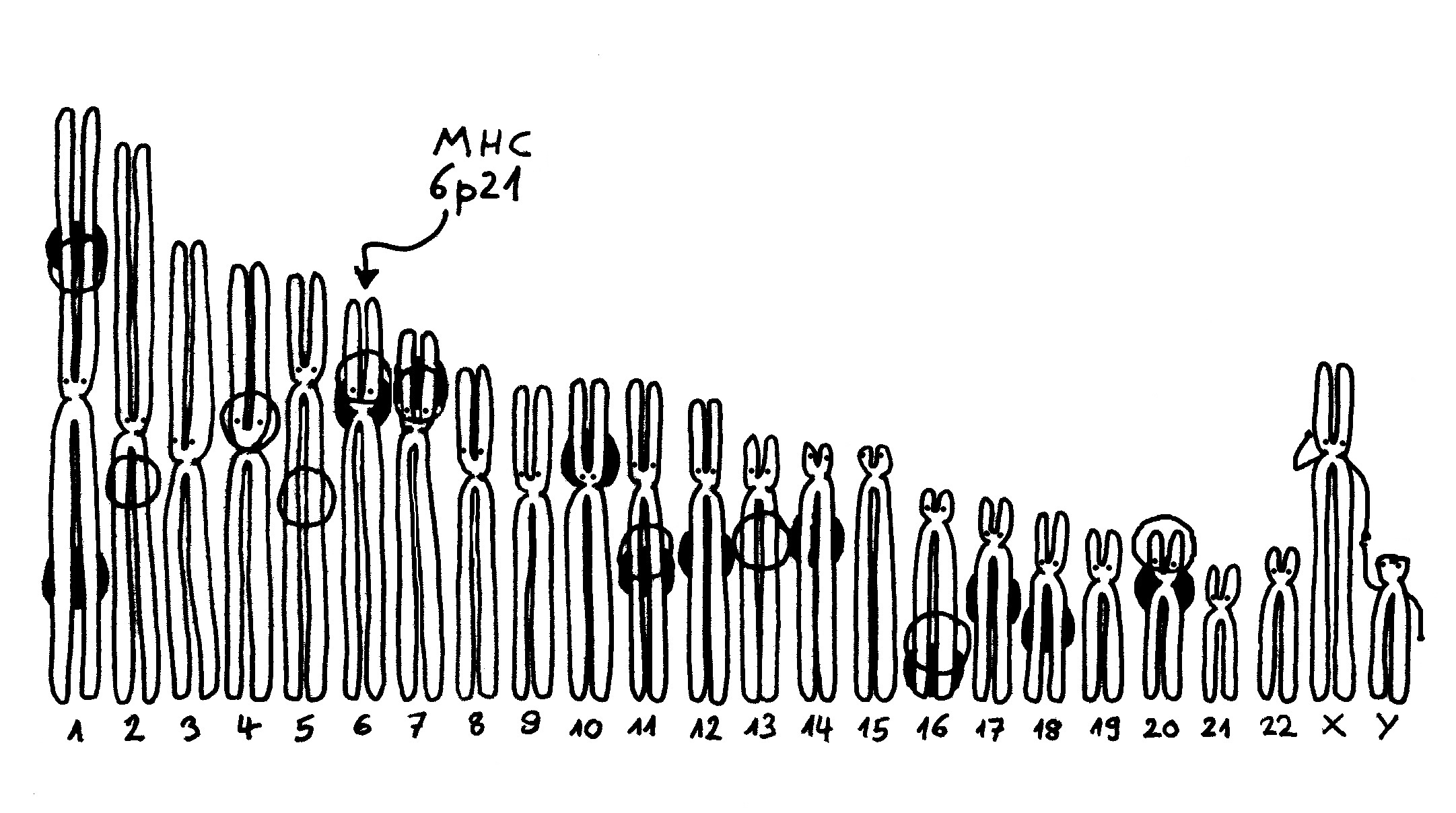
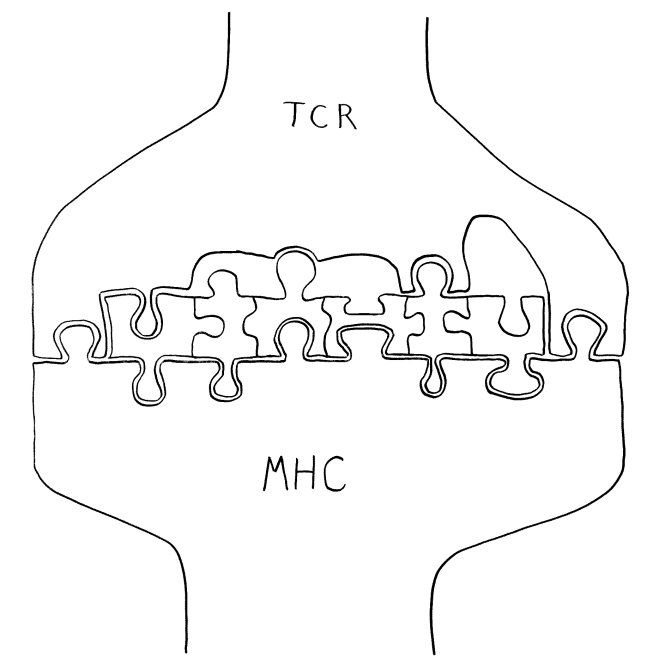
Aber wieso stößt sich das Gewebe des Weibchens und der Männchen nicht ab? Schließlich sind die Tiefseeangler Knochenfische, und diese haben – genau wie wir – neben der angeborenen eigentlich eine erworbene Abwehr. MHC-Klasse-I- und -II-Komplexe, auf denen Antigene präsentiert werden. Zytotoxische T-Zellen, die Eindringlinge töten. T-Helferzellen, die Zytokine ausscheiden. B-Zellen und von ihnen produzierte Antikörper verschiedener Klassen, die fremde Zellen und Fremdkörper markieren und bekämpfen. All diese Komponenten der erworbenen Abwehr sind bei uns Menschen an Abstoßungsreaktionen beteiligt, etwa nach Organtransplantationen.
Ein Forschungsteam um Thomas Boehm hat dieses Rätsel 2020 ansatzweise gelöst. Es hat das Genom von 13 Ceratioidei-Arten analysiert: 3 Arten, bei denen die Männchen nicht mit den Weibchen verwachsen (die Kontrollgruppe), 4 Arten, bei denen die Männchen vorübergehend an ihnen festmachen, 3 Arten, bei denen jeweils ein einzelnes Zwergmännchen permanent mit einem Weibchen verwächst, und schließlich 3 Arten, bei denen sich mehrere permanent festgewachsene Zwergmännchen ein Weibchen teilen.
To make a long story short: Das Immunsystem der Arten, bei denen die Geschlechter eigenständige Orgainsmen bleiben, verfügt über zytotoxische T-Zellen, T-Helferzellen und B-Zellen, die Antikörper herstellen und eine Affinitätsreifung durchlaufen, also bei der Bekämpfung ihrer spezifischen Feindbilder immer effizienter werden. Alles ganz normal für Wirbeltiere, die so Krankheitserreger, Tumorzellen usw. bekämpfen.
Die Arten, bei denen sich die Zwergmännchen phasenweise an die Weibchen anflanschen, haben ebenfalls beide T-Zell-Typen und Antikörper-produzierende B-Zellen; bei ihnen fällt aber die Affinitätsreifung aus, weil die dafür erforderlichen Enzyme mutiert sind. Vermutlich würde diese Optimierung die Antikörper so schlagkräftig machen, dass schon eine vorübergehendes Verwachsen zu Abstoßungsreaktionen führen könnte.
Arten, die permanente Gespanne aus einem Weibchen und einem Männchen bilden, haben ebenfalls keine Affinitätsreifung – und darüber hinaus keine zytotoxischen T-Zellen. Die polyandrischen („vielmännigen“) Tiefseeanglerfisch-Arten schließlich müssen auch ohne T-Helferzellen und ohne jeden Antikörper auskommen: Alle entsprechenden Gene sind durch Mutationen ausgefallen.
Weder in der Forschungsarbeit von 2020 noch in mehreren Kommentaren dazu oder einem Review von 2022 habe ich eine gute Erklärung dafür gefunden, dass T-Helferzellen und Antikörper offenbar beim Verschmelzen eines Weibchens mit einem einzigen Männchen nicht zu einer Abstoßungsreaktion führen, obwohl alle Zellen des Männchens für das Weibchen allograft, also fremdes Gewebe sind – und umgekehrt.
Womöglich sind sich die Männchen der polyandrischen Arten untereinander noch fremder, weil sie miteinander um die Befruchtung der Eier des Weibchens konkurrieren? Das Problem wäre dann gar nicht die drohende Abstoßung durch das Immunsystem des Weibchens, sondern die Abstoßung des einen Männchens durch das Immunsystem des anderen. Aber hier spekuliere ich; hoffentlich löst in den kommenden Jahren jemand dieses Rätsel.
Ebenso rätselhaft ist, wie die Ceratioidei trotz des Verlusts eines großen Teils oder sogar ihrer gesamten erworbenen Abwehr überleben: Wie bekämpfen sie Krankheitserreger, wie handeln sie ohne regulatorische T-Zellen und deren friedlich stimmende Botenstoffe die Symbiose mit den Leuchtbakterien aus? Womöglich haben Teile der angeborenen Abwehr diese Aufgaben übernommen. Aber im Unterschied zu anderen Knochenfischfamilien, denen ebenfalls Teile des erworbenen Immunsystems abhanden gekommen sind, hat man im Genom der polyandrischen Tiefseeangler keine Kompensationsmaßnahmen entdeckt, etwa eine massive Ausweitung der MHC-Klasse-I-Gene. Im Gegenteil: Diese scheinen auch recht spärlich vertreten zu sein.
Offen ist auch die Reihenfolge der Ereignisse: Haben die Fische zuerst durch eine Art genetischen Großunfall Teile ihres Immunsystems eingebüßt – und dann in den folgenden Jahrhunderttausenden das Beste daraus gemacht, nämlich ihr Partnersuche-Problem durch Verschmelzen von Weibchen und Männchen gelöst? Oder war das ein schleichender Prozess der Annäherung der Geschlechter, bei dem immer nur diejenigen Paare überlebten, deren Immunsysteme auf das Gewebe der Partner so schwach wie irgend möglich reagierten? Ein echtes Henne-Ei-Problem, mit dem ich euch nun in die finstere, kalte Tiefseenacht entlasse.
Literatur:
E. Gering. Anglerfish are not sexual parasites (Leserbrief zu Swann et al.)