Notizen noch nicht allgemein verständlich aufbereitet; für Teil 4 (Individualentwicklung Immunsystem) und Teil 5 (Evolution) des Buches:
Schnorr S. L. et al. (2014): Gut microbiome of the Hadza hunter-gatherers. Nature Communications 5: 3654, doi:10.1038/ncomms4654 (Open Access)
Abstract: Erstmals Darmflora ursprünglich lebender Jäger und Sammler analysiert und mit dem Mikrobiom von Italienern sowie Ackerbauern aus Burkina Faso und Malawi verglichen. Mikrobenreichtum und Biodiversität größer als in italienischer Stadtbevölkerung. Einzigartig: keinerlei Bifidobacterium; Unterschiede in der Darmflora von Männern und Frauen; Anreicherung von Prevotella, Treponema und unklassifizierten Bacteroidetes, die vermutlich beim Aufschluss ansonsten unverdaulicher Kohlenhydrate aus der überwiegend pflanzlichen Kost helfen; ungewöhnliche Proportionen bei den Clostridiales.
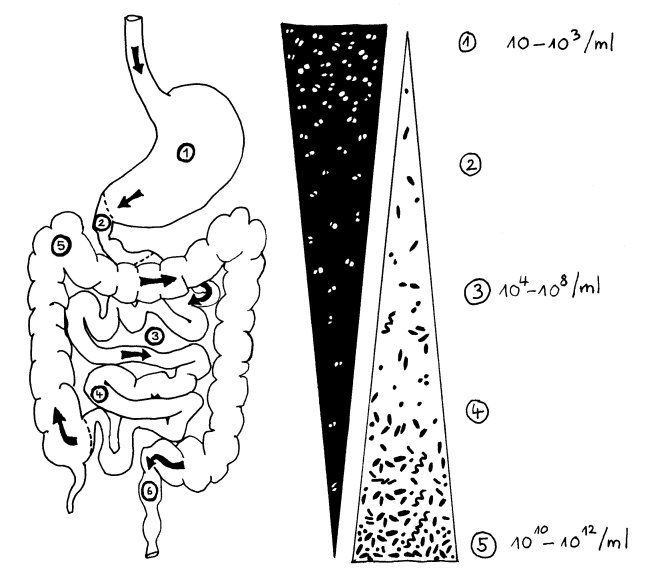
Intro: In Darmflora in ländlichen Gemeinschaften (wenig Antibiotika und „schlechtere“ Hygiene, unraffinierte, saisonal geprägte Kost) Bacteroidetes und Actinobacteria angereichert; in „westlicher Welt“ Diversität und Stabilität des Darm-Mikrobioms verringert. Wissenslücke: Darmflora von Jägern und Sammlern, obwohl das über 95% unserer Evolution unsere Lebensweise war. Hier: Stuhlproben von 27 Hadza aus zwei Lagern analysiert, die zu den etwa 200-300 letzten traditionell lebenden Hadza gehören – einer der letzten Jäger- und Sammler-Kulturen der Welt. Zwar sind sie moderne Menschen, aber sie leben am Eyasisee im Ostafrikanischen Graben in einer Umwelt, die derjenigen unserer Urahnen sehr ähnelt. Vergleich: Darmflora von 16 erwachsenen Italienern aus Bologna und Daten aus Burkina Faso und Malawi. Hadza und Italiener: selbes mittleres Alter (32 J.). Weiterlesen