Dieser Beitrag wird persönlich und am Ende auch emotional. Die medizinischen Sachverhalte erkläre ich nur oberflächlich und nur dort, wo es mir für das Verständnis notwendig erscheint. In nächster Zeit wird es vermutlich auch wieder sachlich-wissenschaftliche Artikel geben, in denen ich zumindest auf einen Aspekt genauer eingehe. Denn durch einen dieser irren Zufälle, die das Leben für uns bereithält, habe ich mich in den Wochen unmittelbar vor den hier geschilderten Vorgängen intensiv mit dem Wirkmechanismus von Glucocorticoiden beschäftigt.
Am 12. November, einem normalen Arbeitstag im Institut, hatte ich nachmittags eine Netzhautablösung. Ich kam aus einer Besprechung, und von jetzt auf gleich erschien im rechten Gesichtsfeld ein dunkelgrauer Viertel-, später dann Drittelkreis, das heißt: Der Bereich, in dem ich nichts mehr sah, wurde rasch größer. Ich erkannte sofort, was los war, da sich meine Netzhautablösung am anderen, linken Auge vor knapp 14 Jahren durch einen ähnlichen Ausfall bemerkbar gemacht hatte. Wie damals sah ich auch diesmal keine Lichtblitze, von denen ja immer die Rede ist als Alarmsignal. Ich bin noch fünf Minuten an die frische Luft gegangen, um auszuschließen, dass einfach mein Kreislauf schlapp gemacht hatte. Als sich die Sicht nicht besserte, brach ich rasch auf, denn eine Netzhautablösung ist ein Notfall, der eine sofortige Reaktion erfordert.
Durch eine veraltete Angabe auf der Website meiner Augenpraxis habe ich mich erst dorthin begeben, um meine Vermutung überprüfen zu lassen. Ich stand vor verschlossener Tür. Gerade habe ich noch einmal nachgesehen: Die falschen Öffnungszeiten stehen immer noch auf der Website, obwohl ich die Praxis sowohl per SMS als auch per Mail auf den Fehler aufmerksam gemacht habe. Also weiter nach Hause, mit der beschissenen Praxis-KI telefoniert und diese angebrüllt, weil sie die Option „Notfall“ nicht vorsieht und mich nicht zu einem Menschen durchstellen konnte, obwohl der Praxisverbund angeblich noch einige Stunden erreichbar sein sollte. Aufgelegt, den Dienst-Laptop ausgepackt, das L-Thyroxin, die Zahnbürste, das Ladekabel und noch ein paar Kleinigkeiten in den Rucksack geworfen, ein Taxi bestellt und mich zur Notaufnahme der Uni-Augenklinik fahren lassen. (Als ich gerade mit dem Taxiunternehmen sprach, versuchte wohl tatsächlich jemand vom Praxisverbund, mich zurückzurufen – zu spät. Als ich im Taxi saß, schickte man mir eine SMS mit der dringenden Empfehlung, mich zur Uni-Klinik zu begeben. Ach.)
Dort das übliche, langwierige, mehrschrittige Aufnahmeverfahren: zentrale Anmeldung, dann (eine Neuerung gegenüber Anfang 2012!) eine Etage tiefer in eine unabhängige Augenarztpraxis, die die Triage vornimmt. Weittropfen, warten, der Arzt kam, ich war als Erste dran, er hatte zwei Botschaften für mich. Erstens ein Lob: Gut, dass Sie so schnell reagiert haben! Zweitens: Ja, das ist eine Netzhautablösung. Überweisung in die eigentliche Uni-Augenklinik. Dort weitere Tropfen, Augendruckmessung, eine weitere Untersuchung, die die Netzhautablösung bestätigte, Ausfüllen eines Selbstauskunftbogens, stationäre Aufnahme. Gespräch mit einer Ärztin, am nächsten Tag sollte ich operiert werden.
Sowohl im Sebstauskunftbogen als auch mündlich wies ich auf meine Glucocorticoid-Unverträglichkeit hin, die sich 2021 nach meiner zweiten Katarakt-Operation gezeigt hatte: Die steroidhormon- bzw. glucocorticoidhaltigen Tropfen, die man mir verschrieben hatte, um eine Entzündung des operierten Auges zu verhindern, ließ nach einigen Wochen meinen Augeninnendruck in sehr ungesunde Höhen steigen. Die Tropfen wurden abgesetzt, der Druck wurde medikamentös gesenkt und in den folgenden Monaten und Jahren immer wieder überwacht – erst engmaschig, nach seiner Normalisierung noch alle sechs Monate. Denn ein zu hoher Augeninnendruck schädigt über kurz oder lang den Sehnerv, was zur Erblindung führen kann. Eine solche Reaktion auf Glucocorticoid-haltige Augentropfen wird als Steroid-Glaukom bezeichnet; sie tritt bei etwa jeder 20. Person auf. Diese Menschen nennt man Steroid-Responder. Warum das so ist und warum 95 Prozent der Bevölkerung bei identischer Behandlung von diesem Effekt verschont bleiben, ist unbekannt.
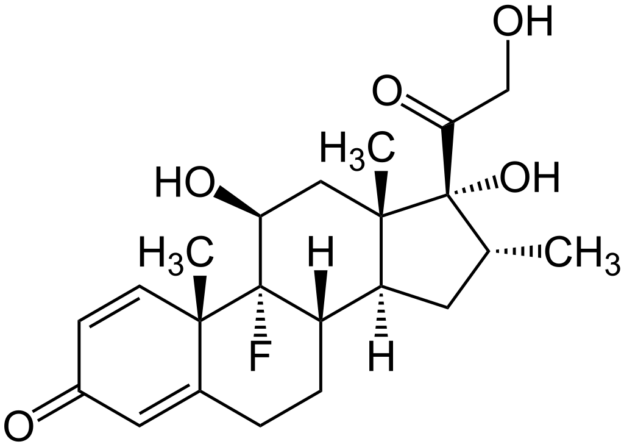
Den Ärzt*innen und Pfleger*innen war diesmal, 2025, also im Prinzip bekannt, was Glucocorticoid-Tropfen in diesem Auge angerichtet hatten. Oder: Es hätte ihnen bekannt sein müssen, wenn sie meine mehrfachen Hinweise ernst genommen hätten. Die Operation verlief normal, abgesehen davon, dass man (wie übrigens auch bei der Star-Operation 2021) ein kleines Blutgefäß getroffen hatte, sodass meine Sklera hinterher blutrot war. Am Tag nach der Operation erhielt ich erstmals Tropfen, und zwar (wie ich jetzt weiß) solche mit dem stärksten Glucocorticoid überhaupt: Dexamethason. Je stärker das Glucocorticoid, desto höher die Wahrscheinlichkeit, dass ein Steroid-Glaukom auftritt. Erneut wies ich auf den Vorfall 2021 hin, aber es hieß: Egal, das wirkt am besten, wir versuchen es damit.
Bei den Messungen in der Klinik und auch bei den beiden ersten Kontrollterminen bei einer niedergelassenen Ärztin in der Woche nach meiner Entlassung war der Augeninnendruck tatsächlich normal. Wie ich inzwischen weiß: kein Wunder, denn nach Beginn der Glucocorticoid-Behandlung dauert es oft Wochen, manchmal Monate, bis der Druck entgleist. Da darf man sich nicht zu früh in Sicherheit wiegen. Genau das ist aber geschehen: Zwischen dem zweiten und dritten Verlaufskontrolltermin in der Praxis lagen 13 Tage, fast zwei Wochen. Und in dieser Zeit – in der ich weiterhin viermal täglich die Glucocorticoid-Tropfen applizierte, außerdem abends eine Salbe mit demselben Wirkstoff – ist es passiert: Der Druck ist völlig entgleist.
Ich merkte davon lange nichts; anders als viele andere Betroffene hatte ich keine Schmerzen. Erst kurz vor dem dritten Kontrolltermin kam es vor, dass mir beim Aufstehen aus dem Sitzen schwarz vorm rechten Auge wurde – komplett, im gesamten Gesichtsfeld, für ein oder zwei Sekunden. Zwei Tage vor der Kontrolle geschah es zum ersten Mal, am nächsten Tag zwei- oder dreimal, beim Kontrolltermin selbst im Wartezimmer noch einmal. Der Sehnerv fuhr durch die abrupte Druckänderung beim Aufstehen kurz komplett herunter; danach war wieder alles „gut“. Die Messungen mit zwei verschiedenen Methoden ergaben dann am Donnerstagmorgen einen Augeninnendruck von 50 mm Hg: ein exorbitant hoher, äußerst gefährlicher Wert, der umgehend durch die Gabe sowohl von Tropfen (BRIMO-Vision sine) als auch einer Tablette (Glaupax) gesenkt werden musste. Einige Stunden später wurde ich erneut vorstellig; der Wert lag da bei 30 mm Hg: Die Medikation wirkte also, reichte aber noch nicht. Abends sollte ich einmalig erneut eine halbe Glaupax nehmen. Die Glucocorticoid-Tropfen wurden selbstverständlich sofort abgesetzt.
Am folgenden Tag, vorgestern, wurde der Druck erneut zweimal gemessen. Morgens war er noch zu hoch, am frühen Nachmittag lag er bei 20, also im Normalbereich, der bis etwa 21 mm Hg reicht. Gestern wurde der Druck in einer anderen Filiale der Augenpraxiskette, die auch am Samstag Patienten empfängt, zu 21 im betroffenen rechten und 20 im linken Auge bestimmt. Das sieht doch ganz gut aus. Dennoch werde ich BRIMO-Vision sine wohl noch eine Weile weiter zweimal täglich tropfen müssen – zusätzlich zu einem anderen Tropfen, mit dem ich mittags das Makula-Ödem bekämpfe, eine weitere postoperative Komplikation, die zu stark verzerrtem, verkleinerten und unscharfen Sehen führt und schon für sich genommen ziemlich lästig und beängstigend sein kann. Und dann ist die Frage, wie sich der Augeninnendruck entwickelt, sobald ich BRIMO-Vision sine absetzen darf. Und wie es um die Netzhaut steht, denn die kann erst per Ophthalmoskopie untersucht werden, wenn die Glaukom-Gefahr gebannt ist.
Das alles klingt vermutlich arg genervt, aber doch noch sachlich. Ich rationalisiere im Allgemeinen ganz ordentlich, lade mir medizinische Fachlieratur zu den Erkrankungen und Fachinfos zu den Medikamenten herunter, höre Hörbücher und Podcasts, schlage mit Spaziergängen im Zoo und im Veedel die Zeit tot, soweit das dunkle, regnerische Wetter es zulässt. Bis gestern war ich zwar energielos, sauer und traurig, aber alles hielt sich im Rahmen. Letzte Nacht jedoch hatte ich einen Albtraum, aus dem ich hochschreckte, und dann überrollte mich die Angst vorm Erblinden. Dem linken Auge geht es nämlich auch nicht gut; es zeigt Anzeichen von Überlastung oder aber … hm … Netzhaut-Problemen. Bitte nicht auch das noch, nicht schon wieder.
Lesen und schreiben kann ich seit dem 12. November nur wenig, radfahren überhaupt nicht. Für meine Psyche ist der Mangel an Gedankenfutter, an intellektueller Betätigung – sei es auf der Arbeit oder privat, etwa in Sachen Autoimmunbuch – fatal, gerade in Kombination mit der Dunkelheit da draußen und dem Mangel an körperlicher Betätigung. Denn selbst wenn das Makula-Ödem nicht wäre und ich wieder ordentlich räumlich sehen könnte, müsste ich alles vermeiden, was den Augeninnendruck hochtreiben kann: bücken, heben, aber auch radfahren oder Yoga oder Gymnastik machen. Trotz Vitamin D spüre ich, wie mir die saisonale Depression, die mich im vorigen Winter vollständig verschont hat, auf die Schulter klopft.
Und ich bin wirklich sauer, eher auf das Gesundheitssystem als auf einzelne Personen. Sowohl in der Klinik als auch in der Praxis arbeiten freundliche, engagierte Menschen, die sich um ihre Patientinnen und Patienten bemühen. Aber im Stress, in der Rödelei gehen offenbar selbst so deutliche Warnhinweise, wie ich sie gegeben habe, komplett unter. Wie kann das sein? Warum hat meine jetzige Ärztin keinen automatischen Zugriff auf meine Patientenakte in der anderen Filiale desselben Praxisverbunds, in die 2021 klipp und klar eingetragen wurde: „V. a. Steroid-Responder“? Warum lagen zwischen zwei Kontrollterminen 13 Tage, mitten in dem Zeitfenster, in dem eine Augeninnendruck-Entgleisung bei einem „High responder“ wie mir typischerweise auftritt? Muss ich jetzt wirklich meine ePA aktivieren, der ich mich bisher aus den bekannten Datenschutz-Gründen verweigert habe? Hätte das überhaupt etwas geändert? Muss ich mir „Steroid-Responder!“ auf die Stirn tätowieren lassen, um mein Augenlicht beim nächsten Mal nicht komplett einzubüßen?
Vor allem: Hat der Sehnerv, hat die Makula, haben die Netzhäute diesen Clusterfuck im Großen und Ganzen gut überstanden? Wird alles wieder gut? Ich werde berichten. Am Dienstag ist der nächste Kontrolltermin.

 Die Frage ist uralt: Wie finden Tauben, die man weit von ihrem Schlag entfernt aussetzt, nach Hause? Mein letzter Stand war, dass es etwas mit magnetischen Partikeln in den Schnäbeln der Vögel zu tun haben könnte, aber diese Hypothese ließ sich offenbar nicht eindeutig bestätigen.
Die Frage ist uralt: Wie finden Tauben, die man weit von ihrem Schlag entfernt aussetzt, nach Hause? Mein letzter Stand war, dass es etwas mit magnetischen Partikeln in den Schnäbeln der Vögel zu tun haben könnte, aber diese Hypothese ließ sich offenbar nicht eindeutig bestätigen.