In der Immunologie entwickeln sich die Techniken und mit ihnen im Idealfall auch die Einsichten so schnell, dass fünf oder gar zehn Jahre alte Arbeiten meist zum alten Eisen gehören. Aber es gibt Ausnahmen. Manches Konzept taucht irgendwann wieder aus der Versenkung auf, in der es verschwunden war, weil es zur Zeit seiner Entstehung nicht überprüft und weiterentwickelt werden konnte. Das gilt zum Beispiel für die Hypothese vom geschichteten oder gestaffelten Immunsystem, der layered immune system hypothesis, die 1989 von Leonore und Leonard Herzenberg aufgestellt wurde.
Die Schichten oder Phasen sind dabei ursprünglich sowohl stammes- als auch individualgeschichtlich zu verstehen. Auch wenn der Name Ernst Haeckel nirgends fällt, schwingt dessen biogenetisches Grundgesetz mit, also die Rekapitulationsregel: „Die Ontogenese rekapituliert die Phylogenese.“ In seiner dogmatischen Form war dieses „Gesetz“ nicht zu halten, und Haeckel hat der Sache mit seinen didaktisch geschönten grafischen Darstellungen keinen Gefallen getan.
Aber nach wie vor gilt: Je jünger ein Embryo, desto weniger spezifische Züge seiner Art trägt er, und desto mehr Züge hat er noch mit ähnlich frühen Entwicklungsstadien entfernt verwandter Arten gemeinsam – Züge, die evolutionär älter sind als die gattungs- und artspezifischen Ausdifferenzierungen der späteren Entwicklungsstadien. Auf das Immunsystem bezogen hieße das zum Beispiel: Die Elemente der evolutionär älteren angeborenen Abwehr bilden sich im werdenden Individuum früher heraus als die Bestandteile der evolutionär jüngeren erworbenen Abwehr.
Schon bei den Herzenbergs und erst recht in den neueren Arbeiten, die sich auf die Hypothese beziehen, steht aber die Ontogenese, die Embryonalentwicklung, im Vordergrund. Die Entwicklung des individuellen Immunsystems wird traditionell als Reifung verstanden: Vor der Geburt ist das System unreif – im Sinne von unterentwickelt oder nicht funktionstüchtig; nach der Geburt reift es durch den Kontakt mit Antigenen aus der Umwelt heran; im Alter erschöpft es sich.
In den letzten Jahren mehren sich aber die Anzeichen, dass das menschliche Immunsystem bereits weit vor der Geburt Funktionen erfüllt – nur eben andere als nach der Geburt. Die Geburt markiert also nicht den Beginn der Aktivität, sondern eine Änderung des Aufgabenprofils, die mit einer Änderung der zellulären Zusammensetzung und der „Gestimmtheit“ des Immunsystems einhergeht: mit dem Rückbau einer Ebene und dem Ausbau einer anderen.
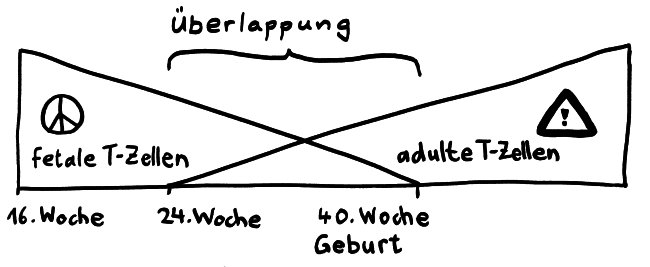
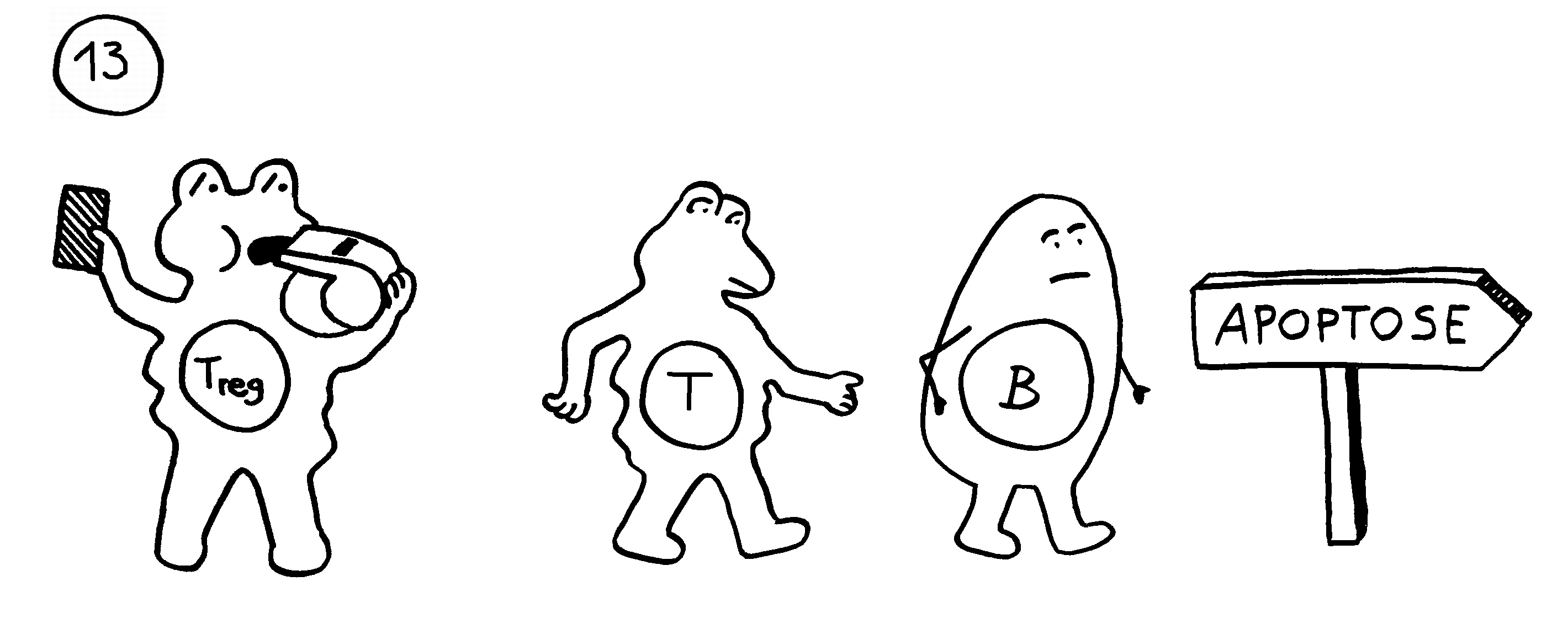
Die Entwicklungsphasen der tolerogenen Immunreaktionen durch fetale T-Zellen und der aggressiven Immunreaktionen durch adulte T-Zellen überlappen sich. Nach Burt 2013, Abb. 1
Die für eine Ebene oder Phase des Immunsystems typischen Lymphozyten besiedeln die Lymphorgane und die Peripherie nicht kontinuierlich, sondern in Wellen. Ein Beispiel sind die beiden B-Zell-Populationen, die bei Mäusen zu unterschiedlichen Zeiten auftauchen, von unterschiedlichen hämatopoetischen Stammzellen im Knochenmark abstammen und unterschiedliche Eigenschaften haben: In neugeborenen Mäusen dominieren die B-1-Zellen, die vor allem in der Bauchhöhle vorkommen; bei erwachsenen Mäusen herrschen B-2-Zellen vor, die schlagkräftigere Antikörper produzieren.
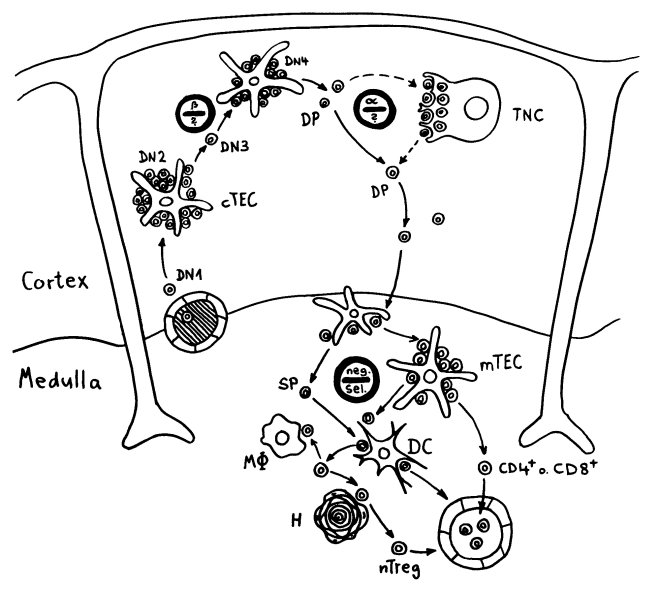
Auch das T-Zell-Repertoire entwickelt sich in Wellen. Wie bereits besprochen, entstehen beim Menschen während der 9. Schwangerschaftswoche zunächst γδ-T-Zellen, die bei Erwachsenen nur noch etwa fünf Prozent der T-Zellen ausmachen. Ab der 10. Woche werden αβ-T-Zellen produziert, und zwar sowohl zytotoxische T-Zellen (CD8+) als auch CD4+-T-Zellen, die entweder zu Helferzellen oder zu regulatorischen T-Zellen (Tregs) werden. Die frühen CD4+-T-Zellen haben eine starke Neigung, sich – manchmal schon im Thymus, zu einem großen Teil aber erst in der Peripherie – zu Tregs zu entwickeln und fortan besänftigend auf das restliche Immunsystem einzuwirken.
Vor allem im zweiten Schwangerschaftsdrittel wimmelt es im Körper des werdenden Kindes von Tregs. In der 24. Schwangerschaftswoche machen sie 15 bis 20 Prozent aller CD4+-T-Zellen aus, während es bei der Geburt nur noch 5 bis 10 Prozent und bei Erwachsenen unter 5 Prozent sind. Fehlen sie, etwa aufgrund eines genetischen Defekts im Treg-typischen Gen FoxP3, so kommt es bereits kurz nach der Geburt zu einer massiven, viele Organe umfassenden Autoimmunreaktion (IPEX). Erst im dritten Trimester werden die tolerogenen fetalen T-Zellen allmählich von aggressiveren adulten T-Zellen abgelöst.
Das kam für viele Forscher überraschend, denn man hatte die Entwicklung der erworbenen Abwehr jahrzehntelang fast nur an Labormäusen erforscht, bei denen die T-Zell-Produktion knapp vor der Geburt anläuft und nicht bereits im ersten Trimester. Die ersten Tregs verlassen den Mäuse-Thymus sogar erst am dritten Tag nach der Geburt. Dieser grundlegende Unterschied zwischen Mensch und Maus ist – wie so vieles – mit der ebenso grundverschiedenen life history der beiden Arten zu erklären.
So, wie das mütterliche Immunsystem während der langen Schwangerschaft beim Menschen vor der Herausforderung steht, den (halb)fremden Fetus nicht abzustoßen, muss auch der Fetus mit (halb)fremden Eindringlingen zurechtkommen, nämlich mütterlichen Zellen und Antikörpern. Mikrochimärismus – der Einbau von Zellen aus der Mutter in den Organismus ihres Kindes ebenso wie der Einbau von Zellen des Kindes in den Organismus seiner Mutter – ist bei Menschen und anderen großen, langlebigen Säugetieren weit verbreitet und in den allermeisten Fällen völlig harmlos: Das Immunsystem lernt rechtzeitig, dass diese Zellen von nun an dazugehören, und die Einwanderer integrieren sich anstandslos. Zu ihnen zählen auch mütterliche Immunzellen aller Art, etwa Monozyten, natürliche Killerzellen, T- und B-Zellen. In den fetalen Lymphknoten präsentieren einige von ihnen den Immunzellen des Kindes mütterliche Antigene.

Die Chimäre der Mythologie ist vorne Löwe, in der Mitte Ziege und hinten Drache. Wir alle sind Chimären: Unsere Körper enthalten Zellklone, die aus unseren Müttern stammen.
Neben mütterlichen Zellen dringen auch mütterliche Antikörper in den Fetus ein, und zwar massenhaft: Gegen Ende der Schwangerschaft ist die Konzentration von mütterlichem Immunglobulin G (IgG) im Fetus höher als im mütterlichen Blut. Über die Muttermilch nimmt das Neugeborene weiter IgG auf. Diese Antikörper schützen das Kind in den ersten Lebensmonaten vor Infektionen. Antikörper sind bekanntlich Proteine und als solche nicht nur Waffen, sondern zugleich Ziele der Abwehr – sofern das Immunsystem nicht lernt, sie zu tolerieren.
Außer mütterlichen Antigenen tauchen währen der Entwicklung des Fetus auch immer wieder neue Gewebstypen und Organe auf und mit ihnen Autoantigene, auf die das Immunsystem nicht aggressiv reagieren darf. Und die bakterielle Flora, die unsere Haut und unsere Schleimhäute unmittelbar nach der Geburt besiedelt, muss zwar in ihre Grenzen verwiesen, aber ansonsten toleriert werden. Ähnliches gilt vermutlich für einige Pathogene, etwa Viren, die die Schutzwälle rings um den Fetus überwinden und ihn bereits vor der Geburt chronisch infizieren können: Auch sie müssen zwar eingedämmt, dürfen aber nicht aggressiv bekämpft werden, weil das für das werdende Kind das Ende bedeuten würde.
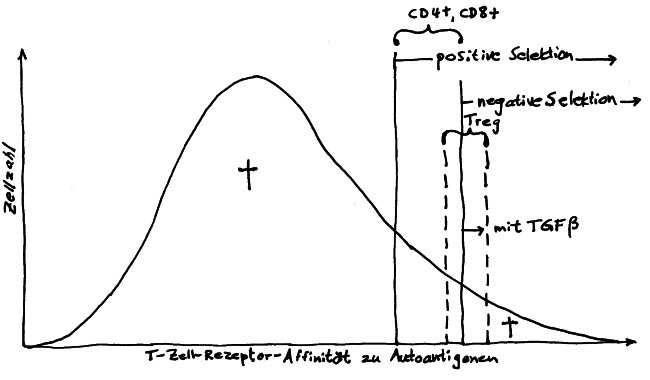
Die zentrale Toleranz durch die negative T-Zell-Selektion im Thymus reicht für diese Herunterregulierung der Abwehr offenbar nicht aus: Auch in der Peripherie muss Frieden gestiftet werden. Naive fetale CD4+--T-Zellen müssen sich bei Bedarf schnell zu antigenspezifischen Tregs weiterentwickeln können. Dazu brauchen sie Signale aus der TGF-β-Familie, die tatsächlich in fetalen Lymphknoten in viel höherer Konzentration vorliegen als in adulten Lymphknoten. Auch können sich fetale Tregs, wenn sie in den Lymphknoten mit Interleukin 2 angeregt werden, stark vermehren, selbst wenn ihre T-Zell-Rezeptoren gerade nicht durch das passende präsentierte Antigen stimuliert werden – was bei adulten Tregs eine strikte Voraussetzung für die Zellteilung ist.
Auch wenn sich fetale und adulte Tregs äußerlich zum Verwechseln ähneln: Sie stammen – wie Experimente an „humanisierten“ Mäusestämmen zeigen – von unterschiedlichen hämatopoetischen Stammzellen ab, haben unterschiedliche Genexpressionsprofile und Aktivierungsschwellen und gelangen in der Peripherie in unterschiedliche Signal-Landschaften, die ihr Verhalten und ihre weitere Entwicklung in entsprechende Bahnen lenken.
Einige Vertreter der Hypothese vom mehrschichtigen oder gestaffelten Immunsystem meinen, die individuell unterschiedliche Neigung zu Autoimmunerkrankungen, Allergien und Nahrungsmittelunverträglichkeiten könne mit dem Mischungsverhältnis zwischen fetalen und adulten T-Zell-Populationen zum Zeitpunkt der Geburt zusammenhängen: Neugeborene, die nur noch wenige fetale, tolerogene T-Zellen aufweisen und dafür bereits sehr viele aggressive T-Zellen vom adulten Typ, könnten im kritischen Zeitfenster nach der Geburt eine bleibende Neigung zu Überreaktionen auf Autoantigene und harmlose fremde Antigene ausbilden.
Die Hypothese vom layered immune system ist nach wie vor umstritten, wie die Diskussion zwischen Mold und Anderson (s. u.) zeigt. Aber sie passt zu den Arbeiten über die Hemmung des bereits voll einsatzfähigen neonatalen Immunsystems durch CD71+-Zellen (junge rote Blutkörperchen), die ich hier vor einigen Monaten in zwei Beiträgen besprochen habe: Offenbar kommen wir – zumindest immunologisch – keineswegs so unreif auf die Welt, wie man früher annahm. Wieder einmal zeigt sich, dass Menschen keine groß geratenen Mäuse sind.
Literatur (chronologisch)
Herzenberg, L. A., & Herzenberg, L. A. (1989). Toward a Layered Immune System. Cell, 59, 953-954. (PDF)
Mold, J. E., & McCune, J. M. (2011). At the crossroads between tolerance and aggression: Revisiting the “layered immune system” hypothesis. Chimerism,2(2), 35–41. http://doi.org/10.4161/chim.2.2.16329
Mold, J. E., & Anderson, C. C. (2013). A discussion of immune tolerance and the layered immune system hypothesis. Chimerism, 4(3), 62–70. http://doi.org/10.4161/chim.24914
Burt, T. D. (2013). Fetal Regulatory T Cells and Peripheral Immune Tolerance in utero: Implications for Development and Disease. American Journal of Reproductive Immunology (New York, N.Y. : 1989), 69(4), 346–358. http://doi.org/10.1111/aji.12083
Loewendorf, A. I., Csete, M., & Flake, A. (2014). Immunological considerations in in utero hematopoetic stem cell transplantation (IUHCT). Frontiers in Pharmacology, 5, 282. http://doi.org/10.3389/fphar.2014.00282
Yang, S., Fujikado, N., Kolodin, D., Benoist, C., Mathis, D. (2015). Immune tolerance. Regulatory T cells generated early in life play a distinct role in maintaining self-tolerance. Science, 2015 May 1;348(6234):589-94. http://doi.org/10.1126/science.aaa7017