#NaNoWriMo22, Tag 3
Im Oktober berichtete Sophie Fessl in The Scientist von einer neu erschienenen Forschungsarbeit aus dem Team um Matthew L. Nicotra. Dem Bericht zufolge sind Immunglobuline wohl viel früher entstanden sind, als man bisher glaubte. Was heißt das genau, warum interessiert mich das, und was ist daran wirklich neu?
Was sind Immunglobuline?
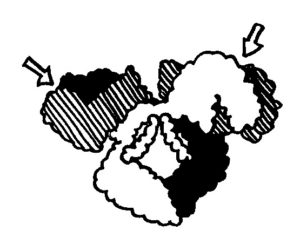
Immunglobuline im engeren Sinne sind Antikörper und B-Zell-Rezeptoren, die wiederum nichts anderes sind als Antikörper mit einem etwas längeren „Stiel“, der sie in der Zellmembran verankert. Diese Proteine dienen der hochspezifischen Erkennung von Antigenen (daher „Anti“), und sie sind kompakt, fast kugelig gebaut (daher „Globulin“, man denke an Globus). Sie enthalten mehrere Immunglobulin-Domänen: Aminosäuresequenzen oder Proteinstrangabschnitte, die sich zu zwei sandwichartig angeordneten blattartigen Strukturen zusammenlagern. Es gibt sogenannte konstante und variable Immunglobulin-Domänen. Die konstanten Domänen bilden zum Beispiel die Stiele der Antikörper, die variablen dagegen die Antigen-Erkennungsstellen (hier mit Pfeilen markiert).
Was ist die Immunglobulin-Superfamilie?
Die Immunglobuline haben eine weitläufige Verwandtschaft: Proteine, die ebenfalls Immunglobulin-Domänen enthalten und allesamt der Oberflächen-Erkennung dienen. Viele von ihnen sind in der Zellmembran verankert und suchen gewissermaßen das direkte Umfeld der Zelle nach passenden Bindungspartnern ab. Zu dieser Großfamilie, der sogenannten Immunglobulin-Superfamilie, zählen zum Beispiel die T-Zell-Rezeptoren, die Haupthistokompatibilitätskomplexe (MHC Klasse I und MHC Klasse II) und etliche Co-Rezeptoren wie CD-8, also viele Proteine, die Funktionen im Immunsystem haben.
Kein Wunder, geht es bei der Abwehr doch oft darum, ganz spezifisch an eine andere Zelle oder ein großes Molekül zu binden, um dieses im nächsten Schritt auszuschalten. Es gibt aber auch andere Gründe, hochspezifisch zwischen eigenen und fremden Zellen zu unterscheiden – eine Fähigkeit, die man Allorecognition, also Fremd-Erkennung nennt. Zum Beispiel kann ein Tier oder Pilz so geeignete Paarungspartner identifizieren, und Blütenpflanzen können eine Selbstbefruchtung, also eine Fusion einer Eizelle mit einer Samenzelle derselben Pflanze verhindern. In diesem Fall ist also – anders als bei der Immunabwehr – das Fremde das Gute und das Identische das Schlechte, denn die sexuelle Fortpflanzung dient ja gerade der genetischen Durchmischung.
Wie lebt das Nesseltier Hydractinia?
Das Forschungsteam um Nicotra untersucht seit längerem ein unscheinbares Nesseltier namens Hydractinia symbiolongicarpus, Die Nesseltiere, zu denen beispielsweise die Quallen und Korallen gehören, haben einen radiärsymmetrischen, z. B. glocken- oder schlauchförmigen Körperbau und nur zwei sogenannte Keimblätter, aus denen sich während der Embryonalentwicklung die Organe und Schichten des Organismus aufbauen. Sie unterscheiden sich also ganz grundlegend von den sogenannten Bilateria oder Zweiseitentieren, die aus drei Keimblättern aufgebaut sind und eine Rücken- und eine Bauchseite haben. Zu diesen zählen beispielsweise wir Säugetiere, aber auch Fische, Mollusken oder Manteltiere. Der letzte gemeinsame Vorfahr der Nessel- und der Zweiseitentiere lebte vor mindestens 600, vielleicht aber auch vor weit über 700 Millionen Jahren – und hatte vermutlich nur zwei Keimblätter.
Hydractinia symbiolongicarpus lebt im Meer und besteht aus einem Geflecht von Röhren, die auf dem Untergrund festgewachsen sind und eine Matte bilden, aus der einzelne sogenannte Hydranthen oder Polypen herausragen, die ein bisschen wie Mini-Seeanemonen aussehen oder wie die Süßwasserpolypen, die wir als Kinder aus Tümpeln gefischt und unter der Lupe beobachtet haben. (Kein Wunder: Beides sind ebenfalls Nesseltiere.) Wie auch bei den Korallen oder Anemonen hat Hydractinia frei schwimmende Larven, die sich in eine festsitzende, mattenbildende Form umwandeln, sobald sie einen passenden Untergrund gefunden haben.
Der Untergrund, auf dem die Matten wachsen, ist typischerweise ein Schneckenhaus – und zwar eines, in dem ein Einsiedlerkrebs lebt. Lebende Schnecken würden eine solche Besiedlung ihrer Gehäuse nicht dulden; den Krebsen ist das egal: Der Bewuchs wiegt nicht viel. Die Nesseltier-Kolonien lassen sich also von den Krebsen herumtragen, filtern mit ihren Tentakeln Nahrung aus dem Meerwasser und wachsen, indem sie am Rand der Matte weitere Röhrchen und Polypen aufbauen.

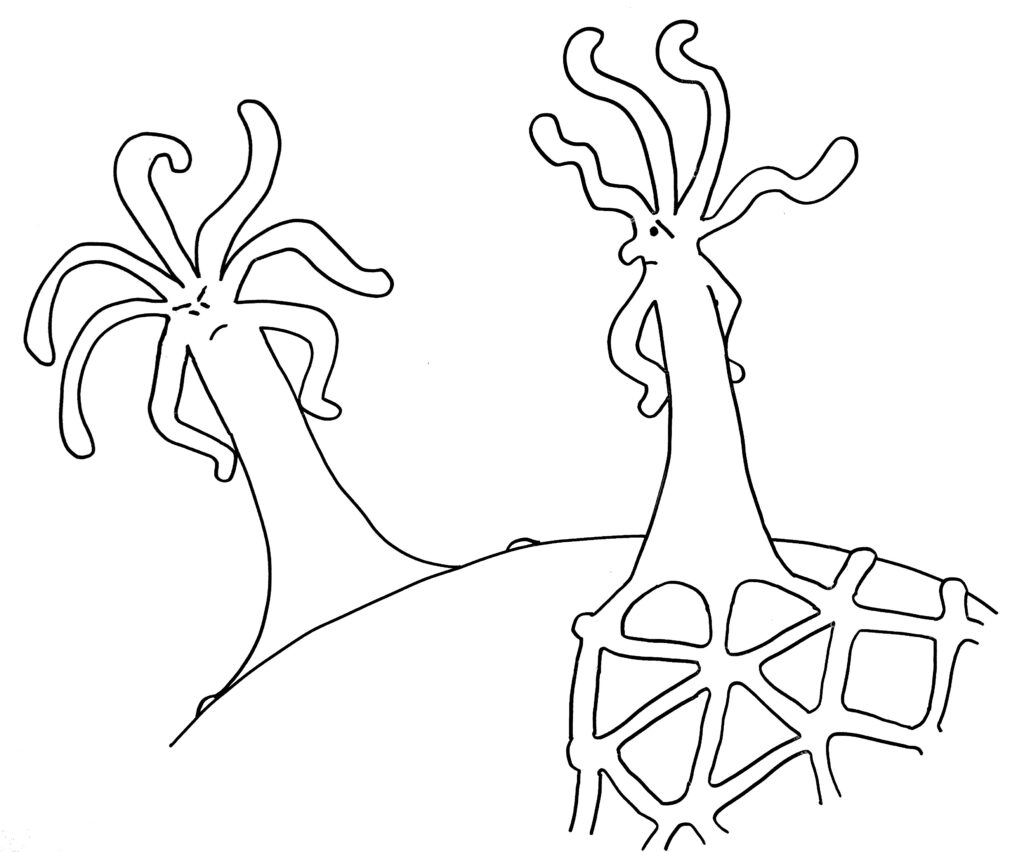
Dabei kann es passieren, dass ein Rand einem anderen begegnet – entweder, weil eine Matte einmal um das Schneckenhaus herumgewachsen ist (Skizze oben), oder weil sich zwei Organismen auf demselben Schneckenhaus angesiedelt haben (Skizze unten).

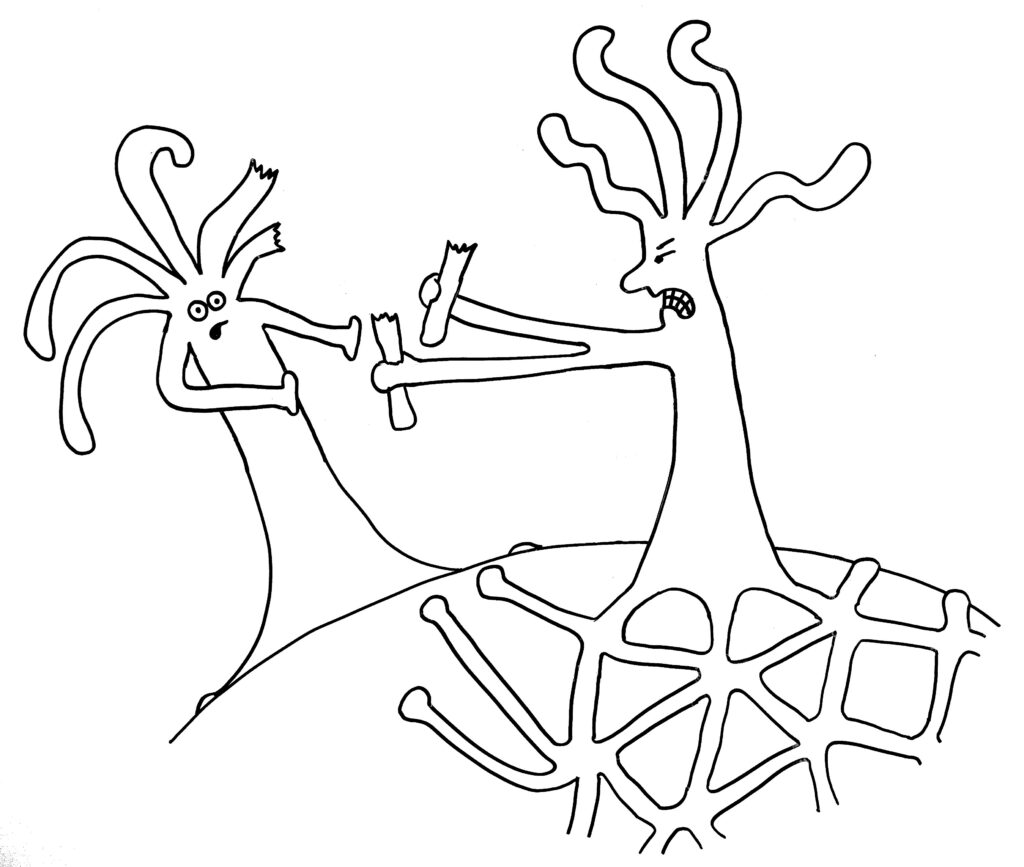
Im ersten Fall verschmelzen die beiden Ränder, im zweiten Fall kommt es nach kurzem „Beschnuppern“ zu einer heftigen Abstoßungsreaktion, die einen der Ränder – wenn nicht gar den ganzen unterlegenen Organismus – zum Absterben bringt.
Wozu braucht es einen Gewebeverträglichkeits-Check?
Hier noch einmal die Abfolge der Ereignisse, bei denen Proteine mit Immunglobulin-Domänen eine Schlüsselrolle spielen, indem sie dem Organismus verraten, ob er da gerade sich selbst oder einem genetisch unterschiedlichen Artgenossen begegnet ist.
Zunächst die Begegnung auf einem noch nicht bewachsenen Teil des Schneckenhauses:
Dann die Reaktion: entweder Angriff …
… oder weitere Annäherung und Verschmelzung mit dem anderen Rand desselben Organismus:
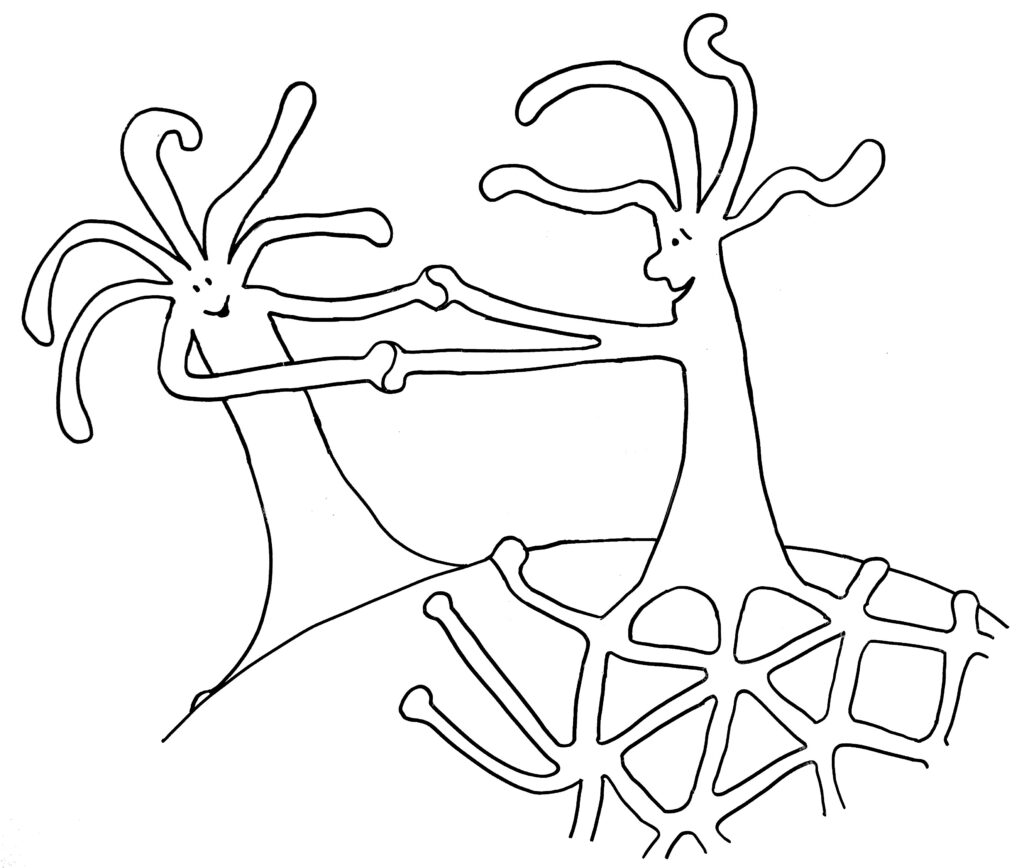
Warum aber ist es den Hydractinien so wichtig, andere Organismen zu bekämpfen? Kann man sich nicht einfach das Schneckenhaus teilen und eine Grenze aushandeln, wie gute Nachbarn? Die Antwort heißt Stamm- oder Keimzellenparasitismus: Wenn sich die Gelegenheit bietet, schmarotzt einer der verschmelzenden Organismen auf den Beiträgen des anderen zum Stoffwechsel und zum Struktur-Aufbau. Er steckt seine Energie ganz in die Produktion von Keimzellen, um seine Gene in der nächsten Generation durchzusetzen – auf Kosten des anderen Organismus, der schuftet, aber kaum Nachwuchs hervorbringen kann. Nur wenn beide Ränder zur selben Kolonie gehören und ihre Zellen dieselben Gene in sich tragen, ist eine Fusion risikolos – ja vorteilhaft, um den begrenzten Platz auf dem Schneckenhaus voll auszuschöpfen.
Haben unsere Gen-Datenbanken blinde Flecken?
Das Forschungsteam hat nun das Genom von Hydractinia symbiolongicarpus komplett sequenziert. Über zwei schon länger bekannte Vertreter der Immunglobulin-Superfamilie hinaus, die banalerweise Allorecognition 1 und Allorecognition 2 heißen, haben sie dabei zahlreiche weitere, ähnliche Gene gefunden. Zusammen bilden sie einen Allorekognitionskomplex (ARC), der an den Haupthistokompatibilitätskomplex (MHC) der Wirbeltiere erinnert. Und wie im MHC sind zumindest einige dieser Gene extrem polymorph; sie unterscheiden sich also von Individuum zu Individuum ein wenig. Zusammen bilden die Immunglobulin-Genvarianten eines Individuums so etwas wie eine eindeutige Personenkennung. Und so, wie diese unterschiedlichen Signaturen bei uns Menschen zu Abstoßungsreaktionen nach einer Organtransplantation führen, lösen sie bei den Hydractinien eine Abstoßung zwischen zwei Organismen aus, die sich auf einem Schneckenhaus begegnen.
Die Forscher*innen waren aber zunächst unsicher, ob die von ihnen entdeckten Proteine wirklich Immunglobulin-Domänen enthalten. Denn die DNA-Sequenzen im Genom und folglich auch die in ihnen codierten Aminosäuresequenzen hatten nur wenig mit den Immunglobulin-Domänen anderer Tiere in den großen Genomdatenbanken gemeinsam.
Erst als das Team von dem Google-Programm AlphaFold die Sequenzen in dreidimensionale Proteinknubbel umrechnen ließ, wurde klar: Ja, das sind wirklich Immunglobulin-Domänen. Wenn wir uns die Proteine als Schlüssel vorstellen, so sind die Datenbanken voll mit Schlüsseln nach dem oberen der beiden folgenden Baupläne:
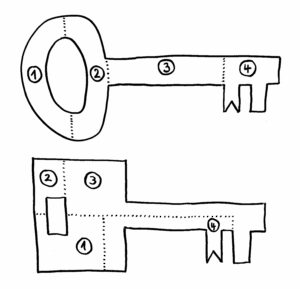
Sieht man sich nur die jeweils vier Bestandteile der Schlüssel an, so erkennt man kaum eine Gemeinsamkeit. Erst wenn man die Teile richtig zusammensetzt (so, wie AlphaFold das mit den Nesseltier-Proteinen gemacht hat), erkennt man, dass beide Gebilde dieselbe Funktion haben: Beide haben vorne einen Bart, der in dasselbe Schloss passt.
Die Autor*innen warnen daher davor, den Gen-Datenbanken blind zu vertrauen, wenn man nach entfernten Verwandten oder Vorformen bestimmter Gene und Proteine sucht: In den Daten sind Organismen, die uns selbst ähneln, stark überrepräsentiert. Spuckt ein Datenbank-Abgleich neu sequenzierter Gene aus nur sehr entfernten verwandten Lebensformen wie den Nesseltieren keinen Match aus, kann man daraus nicht ableiten, dass die Gene nicht auf eine gemeinsame Urform zurückgehen oder die Proteine nicht dieselbe Funktion haben.
Was heißt das für die Evolution des Immunsystems – und der Autoimmunstörungen?
Diese Wirbeltierlastigkeit der Datenbanken macht es schwer, das früheste Auftreten von Neuerungen im Immunsystem zu rekonstruieren. Hinzu kommt, dass Immunglobulin-Domänen unterschiedliche Funktionen übernehmen können, auch solche außerhalb des Immunsystems. Und vielleicht gibt es wirklich keinen gemeinsamen Urahn der Nesseltier- und Wirbeltier-Immunglobulin-Domänen, sondern diese wurden unabhängig voneinander zweimal „erfunden“, weil ihre Gestalt für die Aufgabe der raschen und genauen Unterscheidung zwischen eigenen und fremden Zellen oder Zellprodukten unschlagbar gut geeignet ist.
Die meisten Vertreter der Immunglobulin-Superfamilie in unserem Körper übernehmen Aufgaben in der erworbenen oder adaptiven Abwehr, die erst mit den Fischen vor etwa 500 Millionen Jahren aufgekommen ist. Einige sind aber auch der stammesgeschichtlich älteren angeborenen Abwehr zuzurechnen, etwa Zytokin-Rezeptoren oder Rezeptoren der natürlichen Killerzellen. Insofern ist es nicht unplausibel, dass die Immunglobulin-Domäne schon vor der Aufspaltung zwischen den Nesseltieren und den Zweiseitentieren entstanden ist.
Mich hätte interessiert, ob den Nesseltieren bei der Allorekognition auch Fehler unterlaufen: Kommt es vor, dass eine Hydractnie sich selbst attackiert, weil ihre Immunglobulin-Domänen zum Beispiel falsch gefaltet sind und daher eigenes Gewebe irrtümlich für fremdes halten? Dazu habe ich keine Informationen gefunden. Allorekognitionssysteme sind bei Nicht-Wirbeltieren recht weit verbreitet, aber wie präzise sie arbeiten und ob es bei ihnen Störungen gibt, die unseren Autoimmunerkrankungen ähneln, ist wohl offen. Es kann schon sein, dass solche Pannen vorkommen, denn Nicotra und sein Team haben Indizien dafür gefunden, dass eine Abstoßung nach einer Begegnung zweier Kolonieränder gewissermaßen das Standardprogramm ist, das nur dann abgebrochen wird, wenn die Zellen einander als Teile desselben Organismus erkennen.
Wie tief reichen die Wurzeln?
Als ich mich gestern daran machte, diesen Blogartikel zu schreiben, hat mich eine Information aufgehalten, die die ganze Argumentation im eingangs erwähnten Bericht in The Scientist infrage zu stellen droht: Offenbar haben sogar Hefen, also wirklich nur ganz, ganz entfernt mit uns verwandte Lebewesen, die zu den Pilzen zählen, Erkennungsproteine mit Immunglobulin-Domänen – oder zumindest Domänen, die diesen sehr ähnlich sind. Die Proteine heißen Agglutinine und spielen eine Rolle beim Hefe-Sex, bei dem sich zwei unterschiedliche Fortpflanzungszellen finden müssen: solche, die a-Agglutinin an ihrer Oberfläche tragen, und solche, die α-Agglutinin exprimieren.
Gestern habe ich aufgegeben. Heute scheint mir, dass die Verwirrung nicht nur bei mir herrscht, sondern auch in der Wissenschaft: Was ist ein Immunglobulin im engeren Sinne? Was macht ein vollwertiges Mitglied der Immunglobulin-Superfamilie aus? Und was ist diesen Proteinen bzw. Proteindomänen nur homolog, also ähnlich, aber lediglich entfernt mit ihnen verwandt? Die Grenzen scheinen mir noch nicht endgültig ausgehandelt zu sein.
Und so bleibt bis auf Weiteres offen, ob die stammesgeschichtlichen Wurzeln der Immunglobuline, die sowohl an unserer intakten Abwehr als auch an unseren Autoimmunstörungen maßgeblich beteiligt sind, nun bei den frühen Wirbeltieren, bei den noch früheren ersten Chordatieren, bei den noch früheren ersten Zweiseitentieren, bei den uralten gemeinsamen Vorfahren der Nessel- und der Zweiseitentiere oder sogar ganz am Grunde des gesamten Eukaryoten-Stammbaums liegen.

 #NaNoWriMo22, Tag 4
#NaNoWriMo22, Tag 4