Alles Wichtige über die Biologie der Autoimmunerkrankungen lesen? Keine Chance.
Sie dürfen diese Zeichnung gerne in Folien etc. übernehmen, sofern Sie die Quelle angeben: Dr. Andrea Kamphuis, https://autoimmunbuch.de

Alles Wichtige über die Biologie der Autoimmunerkrankungen lesen? Keine Chance.
Sie dürfen diese Zeichnung gerne in Folien etc. übernehmen, sofern Sie die Quelle angeben: Dr. Andrea Kamphuis, https://autoimmunbuch.de
Im vorigen Beitrag habe ich einen Überblick über die Todesarten von Zellen gegeben. Eine Methode des zellulären Selbstmords, die Pyroptose, stelle ich hier ausführlicher vor.
Der 2001 geprägte Name bedeutet so viel wie „Feuertod“. Die Pyroptose ist ein stark entzündliches Todesprogramm, das vor allem mit Bakterien infizierte Zellen aktivieren, um eine Ausbreitung der Infektion zu verhindern. Der Zelltod wird durch einen Proteinkomplex namens Inflammasom vermittelt. Das klassische Beispiel sind Makrophagen, also professionelle Fresszellen aus der angeboren Abwehr, die von Salmonella typhimuroum, Shigella flexneri, Listerien, Legionellen oder anderen Bakterien befallen sind, die in ihrem Zytoplasma leben. Aber auch Zellen der Darmschleimhaut, die mit Salmonellen infiziert sind, sterben durch Pyroptose und entlassen dabei die Bakterien wieder in den Darm, aus dem sie gekommen sind. So verhindern sie, dass die Salmonellen durch die Darmschleimhaut-Barriere tiefer ins Gewebe eindringen.
Evolutionäres Wettrüsten
Zwischen innerzellulären Pathogenen und ihren Wirtszellen herrscht ein Wettrüsten: Die Keime versuchen mit immer neuen Gift- und Signalstoffen, die Selbstmordprogramme der Zellen entweder zu forcieren und zu ihrer eigenen Verbreitung zu nutzen oder zu unterbinden, um im Verborgenen überdauern zu können. Und die Zellen versuchen die Keime entweder zu verdauen oder auszuhungern – oder sich selbst stillzulegen, um die Vermehrung der Keime und damit die Ausbreitung der Infektion zu verhindern. Da Pathogene diese Strategie zu unterwandern versuchen, verfügen Zellen über mehrere Selbstmordprogramme: Im Fall einer Blockade können sie auf eine andere Todesart umschalten.
An der Pyroptose sind wie an der bekannteren intrinsischen Apoptose Enzyme aus der Caspase-Familie beteiligt. Die Ähnlichkeit der Wirkmechanismen könnte auf die Verwandtschaft von Bakterien und Mitochondrien zurückzuführen sein: Diese Zellkraftwerke, deren Durchlöcherung ein zentraler Schritt der intrinsischen Apoptose ist, sind evolutionär wohl aus innerzellulären Bakterien hervorgegangen. Pathogen-Bestandteile oder PAMPs (bei der Pyroptose) bzw. das Protein Cytochrom C aus den Mitochondrien (bei der Apoptose) lösen den Zusammenbau von Proteinkomplexen namens Inflammasom bzw. Apoptosom aus, die die späteren Schritte der Todesprogramme ausführen.
Ein Ende mit Knalleffekt
Ein Inflammasom besteht typischerweise aus Sensoren oder Rezeptoren für bakterielle Moleküle und andere Zellstress-Signale, dem Enzym Caspase-1 und Adapterproteinen. Die Zusammenlagerung dieser Komponenten im Inflammasom aktiviert die Caspase-1. Das Enyzm zerschneidet dann wohl einige Proteine, die an der Glykolyse – dem Zuckerabbau – beteiligt sind. So wird die Herstellung des Energieträgers ATP unterbunden: Sowohl den Pathogenen als auch der Wirtszelle geht gewissermaßen der Sprit aus.
Außerdem zerschneidet Caspase-1 die Vorformen der Zytokine IL-1β und IL-18, sodass sie aktiviert und ausgeschieden werden, in der Nachbarschaft Entzündungsalarm geben und Immunzellen anlocken können – vor allem Neutrophile, die dann Bakterien bekämpfen, die aus den infizierten Zellen ausgestoßen wurden oder entkommen sind. (Die Neutrophilen selbst können keine Pyroptose durchlaufen; sind gegen diese Form des infektionsinduzierten Selbstmords immun und daher ideale Bakterienbekämpfer.) Die gleichzeitige Freisetzung von Zytokinen, Bakterien, antimikrobiellen Substanzen und Alarmsignalen oder DAMPs – etwa dem kürzlich hier vorgestellten Molekül HMGB1 – sorgt für eine besonders energische Immunreaktion.
Anders als bei der weitgehend still verlaufenden Apoptose entstehen bei der Pyroptose außerdem Poren in der äußeren Membran der Zellen, die daraufhin wegen des osmotischen Drucks anschwellen und schließlich platzen (Lyse). In dieser Hinsicht ähnelt die Pyroptose der Nekrose.
Der Auslöser entscheidet über Tod oder Rettung
Der bloße Zusammenbau eines Inflammasoms und selbst die Aktivierung von Caspase-1 sind aber nicht immer ein Todesurteil für die Zelle: Ein Inflammasom, das in einer frisch infizierten Zelle zusammengesetzt wird, ist etwas anders aufgebaut als eines, dessen Zusammenbau durch Gefahrensignale aus der Umgebung der Zelle initiiert wird, etwa aus infizierten Nachbarzellen. Im ersten Fall wird die infizierte Zelle eliminiert und die Nachbarschaft mit starken Entzündungssignalen geflutet. Im zweiten Fall wird stattdessen ein Reparaturprogramm ausgeführt, bei dem die Zelle nicht stirbt, sondern sich selbst heilt, indem sie durch Autophagie defekte Komponenten und Mikroben abbaut und ggf. undichte Membranen flickt.
Wenn die Strategie der verbrannten Erde fehlschlägt
Normalerweise hilft die Pyroptose dem Organismus, infizierte Zellen und mit ihnen die Keime zu beseitigen. Ein exzessives pyroptotisches Makrophagensterben kann allerdings das Immunsystem schwächen, da es dann zu wenig professionelle Fresszellen und antigenpräsentierende Zellen für weitere Immunreaktionen gibt. Etwas ähnliches geschieht bei einer HIV-Infektion: Die Retroviren nisten sich in ruhenden T-Zellen ein, die daraufhin durch Pyroptose sterben. Die Viren werden dadurch aber nicht ganz eliminiert, sondern weichen in andere T-Zellen aus, die dann durch Apoptose sterben. Der Mangel an T-Helferzellen führt schließlich zu AIDS.
Auch droht eine Sepsis, wenn aus zahlreichen pyroptotischen Zellen große Mengen an Alarmsignalen oder DAMPs austreten. Dann bricht ein sogenannter Zytokinsturm los, bei dem eine Massenausschüttung von Zytokinen zahlreiche Immunzellen anlockt, die ihrerseits massenhaft Zytokine ausschütten. Dieser Entzündungsteufelskreis lässt sich oft nicht rechtzeitig stoppen.
Und spätestens bei „Teufelskreis“ ahnt man es: Auch bei einigen Autoimmunerkrankungen könnte Pyroptose eine unglückliche Rolle spielen, weil bei der Lyse der Zellen Autoantigene freigesetzt werden, was Attacken autoreaktiver Immunzellen auslösen oder verstärken kann. Allerdings konnte man bisher nur in wenigen Fällen Bakterien oder andere Pathogene nachweisen, die sich langfristig in unseren Zellen einnisten, so ständig die Pyroptose anheizen und damit schließlich Autoimmunreaktionen auslösen. Wahrscheinlicher ist es, dass in den Zellen von Menschen mit entsprechender genetischer Disposition auch ohne Infektion als Auslöserreiz gelegentlich Inflammasomen zusammengebaut werden, sodass Caspase-1 in Aktion tritt und zur Ausschüttung entzündungsfördernder Zytokine führt: sozusagen ein falscher Feueralarm, der dann wirklich zu einem Brand führt.
Literatur:
Dave Boucher, Kaiwen W. Chen, Kate Schroder (2015): Burn the house, save the day: pyroptosis in pathogen restriction (PDF)
Katherine Labbé, Maya Saleh (2011): Pyroptosis: A Caspase-1-Dependent Programmed Cell Death and a Barrier to Infection (PDF)
Christopher N. LaRock, Brad T. Cookson (2013): Burning Down the House: Cellular Actions during Pyroptosis
Im vorigen Beitrag ging es um Polly Matzingers danger theory, der zufolge ein Gewebe, das eine Infektion oder einen physikalisch oder chemisch bedingten Schaden erleidet, Gefahren- oder Schadsignale aussendet, die das Immunsystem aktivieren. Solche DAMPs (danger- oder damage-associated molecular patterns) können durch Traumata wie Quetschungen, Verschleiß, Verbrennungen oder Infarkte aus dem Inneren von Zellen austreten, in denen sie sich normalerweise verbergen, und lösen dann im Immunsystem eine sogenannte sterile Entzündung aus. Diese dient der Beseitigung verletzter und toter Zellen und der anschließenden Gewebsreparatur.
Sterile Entzündungen haben vieles mit Entzündungen gemeinsam, die durch Infektionen ausgelöst werden; beispielsweise erkennen dieselben Rezeptoren auf antigenpräsentierenden Zellen (dendritischen Zellen und Makrophagen) sowohl DAMPs als auch PAMPs, also pathogen-associated molecular patterns. Eine sterile Entzündung belastet aber das Gewebe weniger und läuft oftmals ohne volle Einschaltung der erworbenen Abwehr ab: Der Einsatz von antigenspezifischen T- und B-Zellen ist ja nicht nötig, wenn die Schadensursache kein Pathogen ist, das diese Zellen passgenau erkennen, bekämpfen und erinnern müssten.
DAMPs und Autoimmunerkrankungen
Sterile Entzündungen und DAMPs interessieren uns hier, weil sie mit Autoimmunerkrankungen in Verbindung gebracht wurden: Nur bei sehr wenigen dieser Erkrankungen ist eine Infektion als Auslöser nachgewiesen; bei anderen wie Rheuma ist die Beteiligung von Pathogenen umstritten. Bei vielen scheint das Immunsystem aber ganz ohne Mitwirkung eines Keims aus dem Ruder zu laufen und sich gegen körpereigene Strukturen zu richten – und zwar dauerhaft, sofern die angeborene Abwehr doch die erworbene Abwehr zu Hilfe ruft.
Die Auslösung steriler Entzündungen ist nur eine der vielen Funktionen von HMGB1.
Ein wichtiges Alarmsignal mit dem sperrigen Namen HMGB1 spielt offenbar bei der Pathogenese von Lupus (SLE), rheumatoider Arthritis, Sjögren-Syndrom, Vaskulitiden wie Morbus Behçet, Sklerodermie, Typ-1-Diabetes und weiteren Krankheiten eine unglückliche Rolle: Im erkrankten Gewebe oder im Blut der Betroffenen ist seine Konzentration gegenüber Gesunden stark erhöht, und in Tierversuchen lassen sich einige Autoimmunreaktionen durch die Ausschaltung von HMGB1 bremsen.
Gerade wegen seiner Schlüsselrolle beim Anstoßen von Entzündungsreaktionen reguliert unser Körper die örtliche Konzentration und das Aktivitätslevel dieses Moleküls normalerweise strikt, und zwar auf mehreren Wegen. Um diese raffinierte Selbstkontrolle – die checks and balances, die bei Autoimmunerkrankungen offenbar versagen – soll es im Folgenden gehen. Um sie zu verstehen, müssen wir uns zunächst das Molekül genauer ansehen.
Steckbrief
Das 1973 aus Kalbsbries (Thymus) isolierte Protein heißt ausgeschrieben „High mobility group box 1“, weil es sich auf einem Elektrophorese-Gel schnell bewegt. Da diese Eigenschaft weiter nichts zur Sache tut, bleiben wir im Folgenden bei der Abkürzung. Das 215 Aminosäuren lange Protein ist evolutionär stark konserviert; 99 Prozent der Aminosäuren stimmen zwischen Maus und Mensch überein. Normalerweise hält es sich im Zellkern auf. 1999 entdeckte man, dass es nicht nur im Zellinneren vorkommt, sondern insbesondere von Makrophagen auch ausgeschieden wird – und zwar relativ spät in einem Entzündungsprozess, nämlich frühestens acht Stunden nach Aktivierung der Immunzellen. So lange brauchen die Zellen, um das Molekül so zu modifizieren, dass es ausgeschleust werden kann.
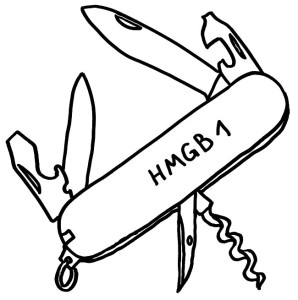
Für diese Ortsverlagerung und den damit einhergehenden Funktionswechsel sind zwei in der folgenden Abbildung mit „Kerntransport“ bezeichnete Abschnitte zuständig, in denen die Aminosäure Lysin angereichert ist. Diese Lysine werden bei Bedarf acetyliert oder phosphoryliert; ihnen werden also Acetyl- oder Phosphatgruppen angehängt. Dank dieses Signals werden sie aus dem Zellkern, in dem sie sonst an die DNA gebunden sind, ins Zytoplasma und ggf. ganz aus der Zelle transportiert.
Das Molekül gliedert sich in drei große Funktionseinheiten: zwei sogenannte HMG-Boxen (A und B), mit denen es sich relativ locker und nicht sequenzspezifisch an DNA anlagert, sowie einen sogenannten C-Schwanz, der bei der Krümmung von DNA zum Einsatz kommt, also etwa bei der Reparatur der Doppelhelix oder beim Ablesen (der Transkription) von Genen. Box B enthält eine Bindungsstelle für den wichtigen Zelloberflächen-Rezeptor TLR4 und an ihrem Ende eine Bindungsstelle für den ebenfalls an Zelloberflächen angesiedelten Rezeptor RAGE. Von beiden wird noch die Rede sein. Weitere Bindungsstellen sind bekannt, hier aber nicht eingezeichnet.
Wichtig sind noch drei Positionen, die mit der Aminosäure Cystein besetzt sind. Deren Seitenketten oder „Reste“ sind – zumindest solange sich das Molekül im Zellkern aufhält – sogenannten Thiolgruppen (-SH), die aus einem Schwefel- und einem Wasserstoffatom bestehen. Ich komme weiter unten auf sie zurück, denn ihre chemische Veränderung bestimmt die Aktivität des Moleküls nach seiner Ausschleusung aus der Zelle.
Je nach Aufenthaltsort unterschiedliche Aufgaben
HMGB1 ist weit mehr als ein DAMP, ein Alarmsignal für antigenpräsentierende Zellen: Außerhalb des Immunsystems übt es zum Beispiel lebenswichtige Funktionen im Nervensystem und im Stoffwechsel aus.
Normalerweise hält es sich im Zellkern auf, wo es als DNA-Chaperon, also als Faltungs- oder Krümmungshelfer für die Doppelhelix dient: Es stabilisiert die dreidimensionale Struktur der Nukleosomen, und es fördert die Transkription der Gene, die somatische Rekombination in B- und T-Zellen, die Reparatur von DNA-Schäden und den Erhalt der Telomere – der Schutzkappen an den Enden der Chromosomen. Kurz: Ohne HMGB1 keine intakten Chromosomen.
Wird es aus dem Zellkern ins Zytoplasma transportiert, was bei Zellstress und vor allem bei einer bakteriellen Infektion der Zelle passiert, fördert es die sogenannte Autophagie, eine partielle Selbstverdauung der Zelle, die den Bakterien die Lebensgrundlage entzieht. Zugleich hemmt es die Apoptose, also den kontrollierten Zelltod.
In die extrazelluläre Flüssigkeit gelangt es entweder durch passive Freisetzung aus allen möglichen beschädigten, sterbenden oder toten Zellen – dann ist es ein Alarmsignal, ein DAMP – oder durch aktive Ausschüttung aus Immunzellen, vor allem aus antigenpräsentierenden Zellen, also Makrophagen und dendritischen Zellen – dann kann man es als Zytokin auffassen, also als Immunsystem-internen Botenstoff.
Es kann als Chemokin dienen, also als Lockstoff für Immunzellen. Es kann andere Immunzellen aktivieren oder deaktivieren, zur Vermehrung bringen und zur Produktion und Ausschüttung von Boten- oder Wirkstoffen anhalten. Es kann alleine agieren, sich aber auch mit anderen Immunzell-Aktivatoren zusammenlagern und dann synergistisch wirken. Es kann als Adjuvans dienen, also Immunreaktionen unspezifisch verstärken. Auch an der Wundheilung im Anschluss an eine Immunreaktion wirkt es mit. Manche Forscher sehen in ihm den zentralen Koordinator oder Choreografen steriler und infektionsbedingter Entzündungen.
Polygamie
Extrazelluläres HMGB1 bindet an mindestens elf verschiedene Transmembran-Rezeptoren, von denen wir hier nur wenige besprechen, nämlich TLR4, RAGE und CD24/Siglec-10. Einige Rezeptoren auf Zellen der angeborenen Abwehr lösen, wenn HMGB1 an ihre Außenseite bindet, innerzelluläre Signalketten wie den NFκ-B-Weg aus, die zur Produktion von entzündungsfördernden Zytokinen führen. Andere Rezeptoren senden dagegen nach HMGB1-Bindung produktionshemmende Signale ins Zellinnere. Darauf komme ich im Abschnitt „Hase und Schildkröte“ zurück.
Es lagert sich auch gerne mit anderen extrazellulären Molekülen zusammen, etwa mit DNA (natürlich!), aber auch mit dem Zytokin Interleukin-1, mit dem Chemokin CXCL12 oder bakteriellen Substanzen wie LPS. Bindet es dann gemeinsam mit ihnen an einen Rezeptor auf einer Immunzelle, so fällt die Wirkung oft stärker oder anders aus als bei einer Bindung des einen oder anderen Partners allein.
So lagert es sich bei einer Bakterieninfektion mit dem bakteriellen Molekül LPS zusammen. Durch das gemeinsame Andocken an einen Rezeptor einer antigenpräsentierenden Zelle wird ein Transportmechanismus ausgelöst, durch den das LPS ins Innere der Zelle gelangt, wo es zu präsentationsfähigen Antigen-Peptiden weiterverarbeitet wird, die dann spezifisch passende T-Zellen aktivieren, sodass die erworbene Abwehr ins Spiel kommt.
Der Rezeptor RAGE ist ebenso polygam wie sein Bindungspartner HMGB1: Zum einen bindet er auch eine Reihe weiterer Moleküle, zum anderen hängt seine Wirkung vom Zusammenspiel mit weiteren Rezeptoren auf denselben Zellen ab. Ein Regulierungsnetzwerk aus HMGB1, seinen extrazellulären Bindungspartnern und seinen Rezeptoren wird rasch unüberschaubar komplex. Konzentrieren wir uns auf drei exemplarische HMGB1-Regulierungsmechanismen: die Freisetzung, den extrazellulären Oxidations-Countdown und das innerzelluläre Wettrennen von Hase und Schildkröte.
Der schlechte und der gute Tod: Nekrose und Apoptose
Um die Logik des ersten Regulierungsmechanismus zu verstehen, benötigen wir Basiswissen über den traumatischen und den programmierten Zelltod, Nekrose und Apoptose genannt. Tatsächlich können Zellen noch auf andere Weisen sterben, etwa durch Pyroptose, ein für innerzelluläre Infektionen typisches Todesprogramm, oder durch NETose, eine Art Selbstmordattentat von Neutrophilen und anderen Granulozyten. Hier beschränke ich mich auf die beiden klassischen Formen des Ablebens. Bei der Nekrose, die unvorhergesehen kommt, schlägt die Zelle leck:

Dabei treten Moleküle wie DNA und allerlei Proteine aus, die normalerweise außerhalb von Zellen nichts zu suchen haben und daher prädestiniert sind, das Immunsystem als DAMP über den Gewebeschaden zu informieren. (Leider können auch T-Zellen und Antikörper auf diese Substanzen überreagieren, wenn sie sie fälschlich als körperfremd auffassen. Das war hier im Blog schon öfter Thema.)
Bei der Apoptose leitet eine Zelle selbst ihren kontrollierten Rückbau und ihre Entsorgung ein, etwa weil sie merkt, dass sie von Viren oder Bakterien befallen, alt oder auf andere Weise geschädigt ist. Sie ruft dazu eine T-Zelle zur Hilfe, die das Rückbauprogramm startet, und ihre Überreste werden von Makrophagen vertilgt:

Bei der Apoptose sollten keine Zellinnereien auslaufen. In der Spätphase kann es aber zu einer sogenannten sekundären Nekrose kommen, bei der das in kleinem Umfang doch passiert. Außerdem scheiden apoptotische Zellen aktiv Substanzen aus, etwa um das Immunsystem über die Ursache ihres Ablebens zu informieren oder um Makrophagen – ihre Totengräber – anzulocken.
Freisetzung von HMGB1: passiv, schnell und reduziert – oder aktiv, verzögert und oxidiert
Bei einer Nekrose tritt HMGB1 passiv aus einer sterbenden Zelle aus, sobald diese undicht wird. Viele nekrotische Zellen haben keine Zeit, das Molekül vorher zu modifizieren. Dann gelangt das HMGB1 in genau der Form in die Gewebsflüssigkeit, in der es im Zellinneren vorlag.
Stirbt eine Zelle dagegen durch Apoptose oder Pyroptose, kann sie das HMGB1 in Ruhe umbauen, um es auf seine künftige Aufgabe vorzubereiten. Zu diesen sogenannten posttranslationalen Modifikationen des Proteins zählen etwa die oben erwähnte Acetylierung oder Phosphorylierung, die für einen geregelten Transport aus dem Zellkern ins Zytoplasma und aus dem Zytoplasma in den extrazellulären Raum sorgen.
Jetzt kommen die drei Thiol-Reste der Cysteine im HMGB1-Molekül ins Spiel, die ich oben im Steckbrief erwähnt habe. Sollte das Schulwissen über Redox-Zustände verschüttet sein, hier eine kurze Auffrischung: Oxidation wurde klassisch als Hinzufügen von Sauerstoff (O wie Oxygenium) oder als Entzug von Wasserstoff (H wie Hydrogenium), also als Dehydrierung aufgefasst. Der umgekehrte Vorgang, also die Hinzufügung von Wasserstoff oder der Entzug von Sauerstoff, wird Reduktion genannt.
Sinnvoller ist es eigentlich, diese sogenannten Redox-Reaktionen als Abgabe oder Aufnahme von Elektronen zu definieren, aber wir bleiben hier beim volkstümlichen Verständnis: Ein frisch aufgeschnittener Apfel hat helles Fruchtfleisch. An der Luft, also bei der Reaktion mit Sauerstoff, oxidiert er. Die Schnittfläche läuft an und wird schließlich richtig braun:

Antioxidantien wie Vitamin C können diesen Prozess aufhalten; sie sind Reduktionsmittel.
Im Normalfall ist das chemische Milieu in einer Zelle reduzierend; es gibt kaum freien Sauerstoff, der HMGB1 oxidieren könnte. Daher liegen im Zellkern alle drei Cystein-Reste als Thiol (-SH) vor. Bei der abrupten Freisetzung aus einer nekrotischen Zelle bleibt das Molekül zunächst in dieser vollständig reduzierten Form.
Gerät das Molekül jedoch in ein oxidierendes Milieu, so verändert es sich schrittweise: Zunächst bildet sich zwischen den beiden benachbarten Resten in Box A eine Disulfidbrücke (-S-S-) aus; die Reste geben also Wasserstoff ab; der dritte Rest liegt weiter als Thiol (-SH) vor. Wird das Molekül weiterhin mit reaktionsfreudigem Sauerstoff konfrontiert, reagieren schließlich alle drei Reste mit ihm und bilden Sulfonsäure (-SO3H).
Diese schrittweise Oxidation kann sogar innerhalb der Zelle ablaufen, wenn sich das chemische Milieu ändert: Manchmal kann das HMGB1 aus nekrotischen Zellen eine Disulfidbrücke ausbilden, bevor es freigesetzt wird. Auch Makrophagen oder dendritischen Zellen, die mit Bakterien infiziert sind und einen programmierten Zelltod namens Pyroptose durchlaufen, enthalten reaktionsfreudigen Sauerstoff und setzen HMGB1 mit einer Disulfidbrücke frei. Bei einer Apoptose wird HMGB1 vor seiner aktiven Ausscheidung oft sogar vollständig oxidiert, denn die Mitochondrien in den sterbenden Zellen setzen bei ihrem Rückbau reaktiven Sauerstoff frei.
Die drei freigesetzten Formen von HMGB1 wirken sich auf das Immunsystem grundverschieden aus, wie ich im nächsten Abschnitt zeige.
Countdown durch Oxidation
Vollständig reduziertes HMGB1 (das mit den drei Thiol-Resten), wie es etwa bei einer Nekrose freigesetzt wird, wirkt als Lockstoff, und zwar in Verbindung mit einem anderen Molekül: Es bindet an das Chemokin CXCL12, das aus aktivierten Immunzellen abgeschieden wird. Das Doppelpack (ein sogenanntes Heterodimer) aus HMGB1 und CXCL12 bindet sehr gut an den Chemotaxis-Rezeptor CXCR4 und führt dazu, dass Immunzellen mit diesem Rezeptor angelockt werden und sich vor Ort vermehren. Findet das reduzierte HMGB1 nicht genug CXCL12-Moleküle vor, kann es durch Bindung an den Rezeptor RAGE sogar die CXCL12-Produktion ankurbeln. Diese Rekrutierung von Immunzellen ist nach einer Nekrose besonders sinnvoll, denn diese Form des Zelltods läuft – wie oben zu sehen war – ungeplant und ohne Beteiligung von Immunzellen ab. Und die Überreste der zugrunde gegangenen Zellen müssen ja beseitigt, die Ursachen bekämpft, die Gewebeschäden repariert werden.
Teilweise oxidiertes HMGB1, das vorne eine Disulfidbrücke und hinten eine Thiolgruppe aufweist, kann dagegen an den Immunzell-Rezeptor TLR4 binden und so die Produktion und Freisetzung von Zytokinen anstoßen, also eine Entzündungsreaktion anheizen. Das ist besonders sinnvoll, wenn bereits Immunzellen vor Ort sind, von denen einige gerade an einer Pyroptose zugrunde gehen: Hier müssen keine weiteren Immunzellen rekrutiert, sondern die anwesenden zur eifrigen Mitarbeit an der Entzündung angeregt werden.
Vollständig oxidiertes HMGB1 hingegen, bei dem alle drei Cystein-Reste als Sulfonsäure vorliegen, kann Immunzellen weder anlocken und zur Vermehrung anregen noch zur Zytokinproduktion bewegen. Es bindet weder an CXCL12 und CXCR4 noch an TLR4: Es trägt überhaupt nichts zu einer Entzündungsreaktion bei, sondern stimmt anwesende Immunzellen tolerant, sozusagen als Anti-DAMP. Diese Wirkung ist besonders sinnvoll, wenn eine Zelle eine geregelte Apoptose durchlaufen hat, denn hier ist keine weitere Immunreaktion nötig: Es reicht, wenn die bereits anwesenden Fresszellen den Rest der gestorbenen Zelle beseitigen.
Zumeist wird HMGB1 also genau in der Form freigesetzt, in der es die jeweils erforderlichen Reaktionen in Gang setzen kann. Analog zum oxidierenden Apfel von oben sind hier die drei Formen des Moleküls dargestellt, ergänzt um die beiden Oxidationsreaktionen durch reaktiven Sauerstoff (ROS) und um die jeweilige Hauptwirkung des Moleküls auf das Immunsystem, hier durch eine T-Zelle vertreten: Anlockung, Aktivierung und Toleranz:
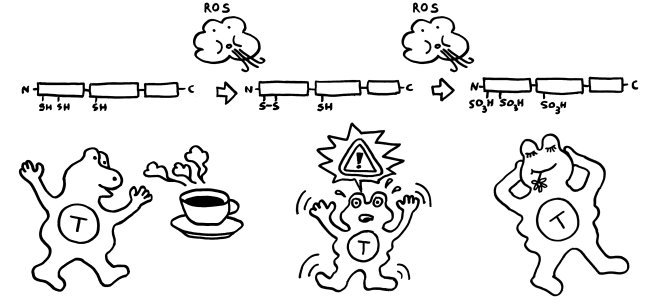
Wie verhindert unser Körper aber, dass das vollständig reduzierte HMGB1, das aus nekrotischen Zellen austritt, immer mehr und mehr Immunzellen rekrutiert und alarmiert, sodass die Entzündung chronisch wird und das umliegende Gewebe Schaden nimmt? Nun: Das extrazelluläre chemische Milieu ist im Vergleich zum gesunden Zellinneren oxidativ. Mit der Freisetzung von vollständig reduziertem HMGB1 beginnt daher ein Countdown. Die extrazelluläre Flüssigkeit oxidiert die Moleküle zunächst teilweise, dann vollständig – bis sie nicht mehr als Alarmsignale wirken.
Hase und Schildkröte
Wir kommen zum dritten Kontrollmechanismus, nämlich der Beschränkung der Zytokinproduktion in Makrophagen und anderen Immunzellen, die HMGB1 in seiner teilweise oxidierten Form aktiviert. Es bindet nicht nur an aktivierende Rezeptoren wie TLR4, sondern auch an inhibierende Rezeptoren wie CD24 und Siglec-10. TLR4 löst daraufhin eine schnelle Signalkette aus, CD24/Siglec-10 eine langsamere:

Beide Signale streben den Chromosomen im Zellkern zu, um dort die Ablesung der Zytokin-Gene zu beeinflussen:

Das vom TLR4 ausgesandte Signal erreicht sein Ziel zuerst und stößt eine starke Produktion entzündungsfördernder Zytokine an:
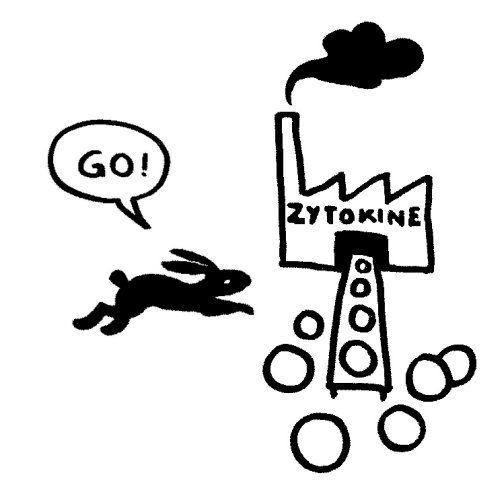
Etwas später trifft das Stoppsignal ein und beendet die Ablesung der Zytokin-Gene. Die Zelle lässt die Produktion auslaufen:
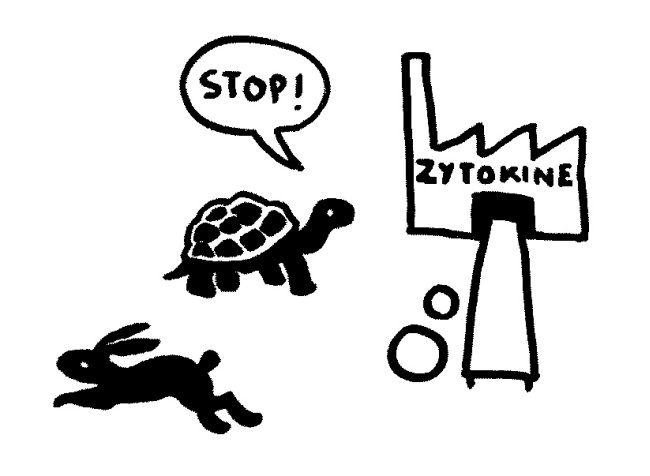
Bei Autoimmunerkrankungen versagen die checks and balances
So raffiniert diese Kontrollmechanismen sind: Bei Autoimmunerkrankungen versagen sie. Welchen Anteil hat HMGB1 an diesen Störungen? Es ist sicher nicht die Hauptursache, aber es kann in Individuen mit entsprechender Veranlagung eine Überaktivierung des Immunsystems fördern. Wichtiger als seine direkte Wirkung auf die Zellen der erworbenen Abwehr, also auf die Vermehrung und Aktivierung von T- und B-Zellen, ist vermutlich die indirekte, nämlich über die antigenpräsentierenden Zellen vermittelte Wirkung.
Sein Beitrag zum Teufelskreis sei am Beispiel Typ-1-Diabetes skizziert: Wenn Betazellen im Pankreas sterben, kann zunächst passiv ein wenig HMGB1 freigesetzt werden. Bei einer unzureichenden Entsorgung der Zellreste reifen durch dieses Alarmsignal oder DAMP zahlreiche dendritische Zellen und Makrophagen heran und werden aktiv: Zum einen können sie selbst noch mehr HMGB1 freisetzen und so die Entzündungsreaktion verstärken. Zu anderen aktivieren sie nun ihrerseits Lymphozyten, indem sie ihnen Antigene präsentieren – unter anderem jene Autoantigene, die zusammen mit dem HMGB1 aus den sterbenden Betazellen ausgetreten sind. In genetisch prädestinierten Individuen greifen diese autoreaktiven Lymphozyten daraufhin Betazellen an, was diese zum Absterben bringt, und so weiter und so fort.
Dieses Modell hat sich im Tierversuch bewährt: Die Pankreas-Lymphknoten von Mäusen mit Diabetes-Neigung, in denen HMGB1 experimentell neutralisiert wurde, enthielten weniger dendritische Zellen des an der Diabetes-Entstehung beteiligten Subtyps und dafür mehr regulatorische T-Zellen (Tregs). In der Milz der Tiere fanden sich zudem sogenannte tolerogene dendritische Zellen, die – wie die Tregs – besänftigend auf Lymphozyten einwirken und so den Teufelskreis durchbrechen können. Es gab zwar noch autoreaktive T-Zellen in den Pankreas-Lymphknoten, aber diese wurden mangels HMGB1 kaum noch in die Langerhans-Inseln gelockt und richteten daher unter den Betazellen wenig Schaden an.
Auch in Versuchstieren mit Lupus-Neigung beteiligt sich HMGB1 an der Pathogenese. Bei ihnen kommen vermutlich zwei Faktoren hinzu: Erstens richtet sich die Autoimmunreaktion bei Lupus primär gegen Zellkern-Bestandteile wie DNA oder Histone – und an diese bindet HMGB1 bekanntlich besonders gut. Es transportiert diese Autoantigene über den Rezeptor RAGE sehr effizient in die antigenpräsentierenden Zellen hinein, was die Autoantigenpräsentation und damit die Aktivierung autoreaktiver Lymphozyten verstärkt. Außerdem tragen auch die Endothelzellen unserer Blutgefäße den Rezeptor RAGE. Bindet dieser HMGB1, so können sich die Blutgefäße entzünden, und es kommt zu einer Lupus-Vaskulitis.
Bei rheumatoider Arthritis scheint zunächst ein Sauerstoffmangel (Hypoxie) Zellen in den Gelenken unter Stress zu setzen. Im Zuge ihrer Stressreaktion setzen sie HMGB1 in die Gelenkflüssigkeit frei, woraufhin sich die Gelenke entzünden können. Das Molekül fördert auch die Neubildung von Blutgefäßen und von Zellen, die Knochengewebe abbauen: sogenannten Osteoklasten. So kann sich die Gelenkschädigung verschlimmern.
Leider ist die Ausschaltung von HMGB1 kein Allheilmittel für Autoimmunerkrankungen. Zum einen hat das Molekül in unserem Körper zahlreiche weitere, lebenswichtige Funktionen. Zum anderen gibt es Hinweise, dass es bei chronischen Entzündungen nicht nur verheerend, sondern auch heilsam wirken kann. So scheint es bei einer Myositis – einer Entzündung von Muskelgewebe – anfangs die Entzündung zu fördern, später aber zum Schutz und zur Regeneration des Muskels beizutragen. Man müsste also eine HMGB1-Blockade entwickeln, die örtlich und zeitlich sehr begrenzt wirkt. Eingriffe in die komplexen und noch nicht vollständig verstandenen checks und balances des Immunsystems bleiben ausgesprochen heikel.
Literatur:
A. Broggi, F. Granucci (2015): Microbe- and danger-induced inflammation
H. E. Harris et al. (2012): HMGB1: A multifunctional alarmin driving autoimmune and inflammatory diseases
R. Kang et al. (2013): HMGB1 in Cancer: Good, Bad, or Both?
S.-A. Lee et al. (2014): The Role of High Mobility Group Box 1 in Innate Immunity
G. Li et al. (2013): HMGB1: the central cytokine for all lymphoid cells
M. Magna, D. S. Pisetsky (2014): The Role of HMGB1 in the Pathogenesis of Inflammatory and Autoimmune Diseases
C. Pilzweger, S. Holdenrieder (2015): Circulating HMGB1 and RAGE as Clinical Biomarkers in Malignant and Autoimmune Diseases
H. Yang et al. (2015): High Mobility Group Box Protein 1 (HMGB1): The Prototypical Endogenous Danger Molecule
J. Zhong (2012/2013): HMGB1 as an Innate Alarmin Promotes Autoimmune Progress: An Essential Role in the Pathogenesis of Type 1 Diabetes
Vor einem Jahr erschien eine Arbeit über das Mikrobiom unkontaktierter Yanomami, die ich damals nur kurz besprechen konnte. Jetzt habe ich sie noch einmal gelesen, obwohl sie immunologisch unergiebig ist: Die Entnahme von Blutproben, die Aufschluss über den Zustand des Immunsystems dieser Menschen hätte geben können, war bei einem Erstkontakt selbstverständlich unmöglich. Man muss schon froh sein, dass sie Abstriche aus ihrer Mundschleimhaut und das Einsammeln von Stuhlproben gestattet haben – vermutlich nicht, ohne sich über dieses merkwürdige Verhalten zu amüsieren.
Die Hauptergebnisse: Die Bakteriengemeinschaften auf der Haut und im Stuhl dieser mutmaßlich seit über 11.000 Jahren isolierten Menschen sind erheblich artenreicher als unsere – und auch als die Mikrobiome anderer naturnah lebender Völker. Die sogenannte Alpha-Diversität ihrer Mikrobiome ist also sehr hoch, vermutlich, weil sie nie mit antimikrobiellen Substanzen zu tun hatten und weil sie in ständigem Kontakt mit ihrer Umwelt leben. In ihrer Darm- und Hautflora leben zum Beispiel Bakterien, die man bislang für reine Bodenbakterien gehalten hat. Zugleich sind die Unterschiede in der Mikrobiom-Zusammensetzung zwischen den 34 Yanomami, von denen die Proben stammen, viel geringer als zwischen denen zweier Menschen aus einer Gruppe aus unserem Kulturkreis. Die sogenannte Beta-Diversität ist mithin sehr klein – wohl wegen des engen Zusammenlebens, der hygienischen Verhältnisse und der gleichartigen Lebensweise und Ernährung aller Gruppenmitglieder.
Unter den Genen dieser Bakterien, und zwar überweigend den Genen von zuvor unbekannten Stämmen des Darmbakteriums Escherichia coli, finden sich 28, die Antibiotika-Resistenzen vermitteln – sogar gegen einige neue, synthetische Antibiotika. Allerdings werden diese Gene in den Bakterien nicht abgelesen, sie sind „stummgeschaltet“ (silenced), sodass die Bakterien anfangs dennoch auf die Antibiotika ansprechen würden. Aber man muss damit rechnen, dass sie sehr bald wirklich Resistenzen entwickeln würden, und zwar gleich gegen mehrere Antibiotika. In Weltgegenden und Kulturen, in denen die sogenannte Therapietreue (die regelmäßige Einnahme des Medikaments über den kompletten notwendigen Zeitraum) vermutlich gering ist, geht das umso schneller.
Erstkontakt: Es gibt keinen Weg zurück
Dem Forscherteam war bewusst, dass die Probensammlung beim Erstkontakt eine einmalige Gelegenheit ist, ein Mikrobiom-Archiv anzulegen, das vermutlich große strukturelle und funktionale Ähnlichkeiten mit dem Mikrobiom unserer altsteinzeitlichen Vorfahren hat – auch wenn sich die einzelnen Bakterien-Arten und -Stämme natürlich auf dem Weg ihrer Wirte nach und durch Südamerika weiterentwickelt haben. 11.000 Jahre entsprechen ungefähr 100 Millionen Bakteriengenerationen. Zugleich begann mit dieser Begegnung zwischen der bislang isolierten Dorfgemeinschaft und den Medizinern und Wissenschaftlern unwiderruflich der Niedergang dieser Diversität – spätestens mit der ersten Antibiotika-Gabe.
Die Autoren schreiben in ihrer Danksagung: „Wir sind auch den Leuten in dem neu kontaktierten Dorf dankbar für ihr Vertrauen und für unser gemeinsamen Wunsch, dass der unvermeidliche Kontakt mit unserer Kultur ihrem Volk gesundheitliche Vorteile und Schutz bringen möge.“ Ist das nicht ein arg frommer Wunsch angesichts der bisherigen Erfahrungen mit der gesundheitlichen und sozialen Entwicklung neu kontaktierter, kleiner indigener Gruppen? Weiterlesen
Es geht, wie so oft, um Ressourcen-Allokation. Wir können jede Kalorie nur einmal ausgeben: zum Nachdenken, für die Vermehrung, im Dienste der Abwehr – am besten dort, wo sie im Moment am dringendsten benötigt wird. Und wenn gerade alles im Lot ist, lagern wir sie ein für kommende Notlagen.
Wohin die Energie fließt, das regelt der Stoffwechsel oder Metabolismus. Er umfasst sowohl biochemische Reaktionswege, auf denen einfachen Rohstoffe unter Energieeinsatz zu komplexeren Strukturen aufgebaut werden, als auch Pfade, auf denen komplexe Biomoleküle zu einfachen Komponenten zerlegt werden, wobei Energie frei wird. Kurz: Metabolismus = Anabolismus + Katabolismus. Damit sich diese Prozesse nicht in die Quere kommen, laufen sie oftmals in getrennten innerzellulären Räumen oder zu unterschiedlichen Zeiten ab.

Action und Substanz: Teile des Zellstoffwechsels machen aus dem Zucker Glukose Energiewährung wie ATP. Andere Zweige des Stoffwechsels produzieren Protein- und Lipidbausteine wie Amino- oder Fettsäuren.
Energie ist eine knappe Ressource; jede Investition in einen Lebensbereich wird mit einem Mangel in einem anderen Bereich erkauft. Das gilt zum einen für ganze Organismen und ihre Organe, etwa für Guppies. Ein Forscherteam hat einen Stamm dieser Aquarienfische über einige Generationen hinweg auf besonders große und besonders kleine Gehirne hin selektiert und dann die Stärke der Immunreaktionen auf transplantierte Guppy-Schuppen gemessen: Die angeborene Abwehr wird schwächer, wenn mehr Energie in die Ausbildung und den Unterhalt eines großen Gehirns fließt. Die erworbene Abwehr bleibt dagegen gleich stark (A. Kotrschal et al., 2016, PDF).
Das gilt aber auch für einzelne Zelltypen wie Tumorzellen oder die Zellen des Immunsystems, die mit Krebszellen einiges gemeinsam haben – etwa die Fähigkeit zur raschen Vermehrung, für die in kurzer Zeit viel Energie benötigt wird. Die Energiequelle ist Glukose oder Traubenzucker, der aus dem Blut in die Zellen gelangt. Naive, d. h. noch nicht mit einem passenden Antigen konfrontierte T-Zellen haben zunächst einen niedrigen Energieumsatz. Sobald sie aber ein zu ihren Rezeptoren passendes Antigen präsentiert bekommen und dadurch aktiviert werden, geht es los: Sie müssen sich massiv vermehren, u. U. weit und mühsam an ihren Einsatzort wandern und eine Menge Wirkstoffe wie Zytokine herstellen. Anschließend leben einige von ihnen als sogenannte Gedächtniszellen noch Jahre bis Jahrzehnte weiter, um bei einem erneuten Auftreten desselben Antigens, also der Rückkehr derselben Gefahr, sehr schnell wieder aktiv zu werden.
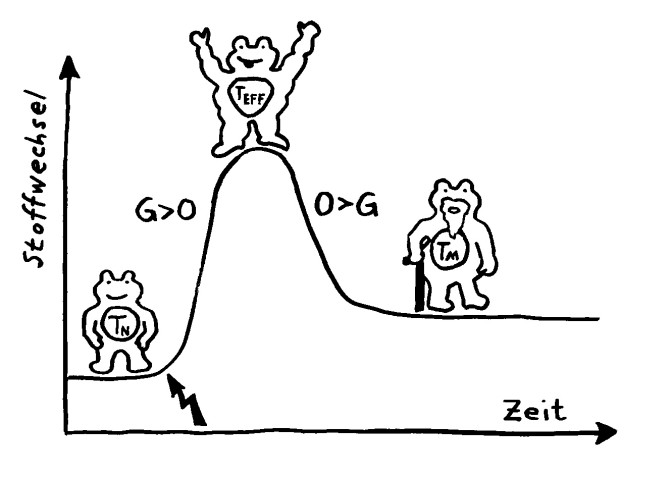
Wird eine naive T-Zelle durch ein Antigen aktiviert (Blitz), kurbelt sie die Glykolyse (G) an, um als Effektor-T-Zelle schnell schlagkräftig zu werden und sich zu vermehren. Als langlebige Gedächtniszelle (M für memory) setzt sie sie danach stärker auf die oxidative Phosphorylierung (O).
Ihr Stoffwechsel passt sich dem Bedarf in diesen drei Lebensphasen an, wobei jede T-Zell-Subpopulation (etwa CD4+, CD8+ oder Treg) ein etwas anderes Programm verfolgt.
Im Ruhezustand gewinnen die naiven T-Zellen Energie aus allen möglichen Quellen, nämlich Glukose, Fettsäuren und Aminosäuren, und zwar größtenteils in ihren Mitochondrien, den Kraftwerken unserer Zellen. Die darin ablaufenden Stoffwechselwege heißen Citratzyklus und oxidative Phosphorylierung, kurz OXPHOS. Sie sind sehr effizient, liefern also sehr viel von dem Energieträgermolekül ATP – das aber recht langsam: ideal für ruhende T-Zellen, die gemächlich durch die Blutgefäße und die Lymphknoten patrouillieren und auf die Präsentation eines Antigens warten, das zu ihren Rezeptoren passt.
Bei ihrer Aktivierung schalten die T-Zellen auf einen als Glykolyse bezeichneten Stoffwechselweg um, der stattdessen im Zellplasma abläuft und Glukose abbaut, um daraus möglichst rasch ATP und die einfachen Grundbausteine Pyruvat und Lactat zu gewinnen. Aus diesen Zwischenprodukten wird dann Zellsubstanz aufgebaut (im Wesentlichen Nukleinsäuren, Fette und Proteine) und die Zellteilung sowie die Wirkstoffproduktion angetrieben. Die Glykolyse hat eine schlechtere Energiebilanz als die Stoffwechselwege in den Mitochondrien, aber dafür ist sie schnell – und auf Tempo kommt es an, wenn eine T-Zelle ihr passendes Antigen erkannt hat und sich rasant vermehren muss, um die Gefahrenquelle zu bekämpfen, bevor der Körper großen Schaden nimmt.
Gedächtnis-T-Zellen sind dagegen wieder auf den Citratzyklus und OXPHOS angewiesen, denn sie müssen sehr lange überleben, um als Archiv für ehemalige Infektionen und andere überstandene Gefahren zu dienen. Sie müssen aber, solange sie nicht reaktiviert werden, kaum Immunsystem-Wirkstoffe herstellen oder einlagern, können also Aminosäuren und Fettsäuren aus nicht mehr benötigten Proteinen und Lipiden ruhig abbauen bzw. in Energieträgermoleküle umwandeln.
Ein Forscherteam um Zhen Yang ist 2015 der Frage nachgegangen, ob die autoreaktiven T-Zellen, die bei Autoimmunerkrankungen auftreten, womöglich einen charakteristisch veränderten Zellstoffwechsel aufweisen. Ihre Idee: Eine Stoffwechselstörung, etwa eine ständige Überproduktion von Energie, könnte die Immunzellen chronisch überaktiv machen – und eine chronische Entzündung unter Beteiligung autoreaktiver T-Zellen ist für Autoimmunerkrankungen typisch, etwa für rheumatoide Arthritis (RA) oder systemischen Lupus erythematodes (SLE). Dann könnte man diese Erkrankungen womöglich durch Eingriffe in den Stoffwechsel der T-Zellen bremsen oder gar heilen.
Das wäre natürlich zu schön gewesen. Leider stellt sich die Lage komplexer dar: Sowohl bei RA als auch bei SLE ist der Stoffwechsel der T-Zellen verändert – aber nicht gleichartig.
Bei RA fahren frisch stimulierte CD4+-T-Zellen die Glykolyse nicht so schnell hoch wie bei Gesunden; sie produzieren nicht so viel ATP und Lactat, teilen sich aber trotzdem lebhaft. Die Bremse ist ein Glykolyse-Enzym mit dem furchteinflößenden Namen 6-Phosphofructo-2-Kinase/Fructose-2,6-Bisphosphatase 3, das wir zum Glück PFKFB3 nennen dürfen. An diesem Enzym herrscht in den T-Zellen von Rheumatikern Mangel, da das entsprechende Gen viel zu schwach abgelesen wird. Die Zwischenprodukte, die sich vor diesem Nadelöhr in der Glykolyse anstauen, weichen auf einen anderen Stoffwechselweg aus: den Pentosephosphatweg. Das führt zu einem Mangel an sogenannten reaktiven Sauerstoffspezies (ROS). Ein ROS-Mangel wiederum geht mit starken Gelenkentzündungen einher; ROS schützt vor Arthritis.
Warum das Enzym PFKFB3 nicht richtig abgelesen wird, ist unklar. Die T-Zellen von RA-Patienten altern vorzeitig. Aber ob diese zelluläre Frühvergreisung durch Energiedefizite aufgrund des Enzymmangels zustande kommt oder umgekehrt das Enzym nicht richtig abgelesen wird, weil die Zellvergreisung das Erbgut schädigt und die Gen-Expression beeinträchtigt, weiß man nicht. Jedenfalls sterben T-Zellen, die nicht genug ATP produzieren, vorzeitig ab. Der dadurch drohende Lymphozyten-Mangel (Lymphopenie genannt) zwingt den Organismus, die Produktion neuer naiver T-Zellen zu beschleunigen. Das geschieht bei älteren Erwachsenen nicht etwa im Thymus, der sich ja bereits zurückgebildet hat, sondern durch verstärkte Teilung der schon im Körper kreisenden naiven T-Zellen: die sogenannte homöostatische T-Zell-Proliferation. Bei diesem Prozess scheinen sich autoreaktive T-Zellen bevorzugt zu vermehren, was zu einer Autoimmunerkrankung führen kann.

Homöostatische T-Zell-Proliferation: Das Repertoire der naiven T-Zellen mit unterschiedlichen Rezeptoren (oberste Reihe: drei Zellklone) bleibt normalerweise bis ins Alter erhalten, weil Verluste durch Teilung der übrigen Zellen kompensiert werden. Bei einer Lymphopenie, also dem massenhaften vorzeitigen Sterben von T-Zellen, wird die homöostatische Proliferation verstärkt. Dabei können Klone verloren gehen (weiß) und autoreaktive T-Zellen (schwarz) sich so stark vermehren, dass eine Autoimmunerkrankung ausbricht.
Auch die T-Zellen von Lupus-Patienten haben einen auffälligen Stoffwechsel. Aber sie produzieren ihr ATP primär auf dem OXPHOS-Weg in den Mitochondrien, nicht durch Glykolyse. Sie produzieren mehr ROS als normale T-Zellen, nicht weniger. Ihre Energiegewinnung ist gestört; sie bauen weder Glukose noch Fettsäuren noch Aminosäuren so effizient ab wie normale T-Zellen. Vor allem freie Fettsäuren häufen sich wegen des gestörten Abbaus an. Der gestörte Fettstoffwechsel wirkt sich auch auf die Fähigkeit der T-Zell-Rezeptoren zur Wahrnehmung von Antigenen aus: Die Zellmembranen von SLE-Patienten enthalten übermäßig viele Glycosphingolipide, also Lipide mit außen anhängenden Zuckermolekülen. Diese speziellen Lipide lagern sich in der ansonsten nahezu flüssigen Zellmembran gerne zu festeren Regionen zusammen, sogenannten Lipid-Flößen, in die wiederum viele T-Zell-Rezeptoren eingebettet sind. Wohl daher nehmen die T-Zellen von Lupus-Patienten besonders leicht Autoantigen-Signale wahr und aktivieren dann ihrerseits B-Zellen, die Autoantikörper herstellen.
Was lehren uns diese gegensätzlichen Stoffwechseldefekte von T-Zellen bei zwei wichtigen Autoimmunerkrankungen aus dem rheumatischen Formenkreis? Dass die Erkrankungsmechanismen ganz verschieden sein können, auch wenn es sich in beiden Fällen um chronische Entzündungen handelt, bei denen das Immunsystem körpereigenes Gewebe angreift. Dass es daher vermutlich nicht das eine Heilmittel geben und überhaupt noch lange dauern wird, bis wir Autoimmunerkrankungen heilen können. Aber auch, dass man vor lauter Botenstoffen, Signalkaskaden und Erbinformationsableserei den Energiehaushalt des Immunsystems nicht außer Acht lassen darf: Das ist nicht etwa reine Information, die da zwischen und in den Zellen weitergeleitet wird. Es sind vielmehr Substanzen, deren Herstellung und Beseitigung zur rechten Zeit, am rechten Ort und in der rechten Menge Kraftakte und logistische Meisterleistungen des Zellstoffwechsels sind.
Heidrun Schaller hat mir vor einigen Wochen ein Exemplar ihres Buchs „Die Paleo-Revolution“ zukommen lassen. Herzlichen Dank dafür – und für die nette Erwähnung in der Danksagung! Höchste Zeit, das Buch kurz vorzustellen.

Eines vorab: ich ernähre mich nicht nach Paleo oder irgendeinem anderen Prinzip, sondern bin Ottonormal-Omnivore. Aber die sogenannte Steinzeitkost interessiert mich, weil sie sich an unserem genetischen Erbe orientiert und weil offenbar etliche Menschen mit Autoimmunerkrankungen gute Erfahrungen mit dieser Ernährungsweise gemacht haben, die sich vor allem durch den Verzicht auf Zucker, Getreide und Hülsenfrüchte und eine relativ fett- und proteinreiche Kost auszeichnet.
Ungefähr seit Ausbruch meiner Autoimmunerkrankung habe ich immer wieder mal mit Verdauungsproblemen zu kämpfen, die weit über das normale Maß hinausgehen. Ich weiß nicht, ob da ein ursächlicher Zusammenhang besteht. Zum Glück berappelt sich mein System bisher immer wieder – aber wenn das noch schlimmer wird und meine Gesundheit und mein Sozial- und Berufsleben ernsthaft beeinträchtigen sollte, werde ich mich vielleicht doch einmal an Paleo heranwagen. Mit Heidruns Buch habe ich dann den idealen Einstieg zur Hand.
Was mich als Naturwissenschaftlerin gleich begeistert hat, ist die sachliche, stets mit Literaturangaben unterfütterte Darstellung – etwa unseres Verdauungssystems, der Ernährung indigener Völker oder des Mikrobioms. Mir ist auch kein anderer „Ernährungsratgeber“ bekannt, in dem gleich zu Anfang der Unterschied zwischen Korrelationen und Kausalzusammenhängen, die Fallstricke von epidemiologischen Studien oder der Publication Bias so verständlich erklärt werden. Überhaupt liest sich das Buch sehr angenehm, sowohl wegen des guten Stils der Autorin als auch wegen des aufgeräumten Layouts.
Die Skepsis der Autorin gegenüber Ernährungsdogmen macht auch vor Paleo selbst nicht halt: Stets ist von einer „Hypothese“ die Rede – allerdings einer besonders plausiblen, eben aufgrund der Orientierung an der Ernährung und Lebensweise unserer Urahnen, denen wir genetisch noch sehr nahe stehen. Im Kapitel „Was unser Körper mit Essen macht und warum er Paleo mag“ wird unter anderem der chemische Aufbau von Fett, Kohlenhydraten und Proteinen sehr anschaulich erläutert, und es wird deutlich, dass eine kohlenhydratarme Ernährung keineswegs zu Mangelerscheinungen oder zur Verfettung führen muss. In der „Anthropologischen Rundumschau“ stellt Heidrun die traditionelle Ernährung verschiedener indigener Völker vor, die je nach den Umweltbedingungen ganz unterschiedlich ausfiel. Entsprechend viele Spielarten kennt auch die Paleo-Ernährung. Außerdem wird deutlich, dass es bei Paleo nicht darum geht, genau „wie in der Steinzeit“ zu leben: Alle Gemüsesorten, die wir heute essen, sind kultiviert, und nahezu alle Fleischlieferanten sind Zuchttiere. Es geht um praktikable Näherungen – wie etwa die Beschränkung auf Fleisch und Butter von Weiderindern anstelle von sojagefütterten Rindern aus der Massentierhaltung.
Im Kapitel „Das Mikrobiom“ erhalten viele Leserinnen und Leser vermutlich zum ersten Mal einen guten Überblick über unsere „Mitbewohner“, ohne die wir unser Essen nicht aufschließen und Krankheitserreger nicht richtig abwehren könnten. Das dicke Kapitel „Paleo trifft auf Entzündung“, in dem unter anderem Autoimmunerkrankungen und Übergewicht behandelt werden, lese ich im Augenblick. Bereits überflogen habe ich das Kapitel „Wie passt Paleo in unsere Zeit?“, in dem sich Heidrun unter anderem mit den Argumenten von Veganern gegen den Verzehr tierischer Produkte auseinander setzt. So verständlich das angesichts der heftigen und dogmatischen Diskussionen in vielen Foren ist: Mich hat das weniger angesprochen als die übrigen Teile des Buches; einige Passagen klingen sehr nach Rechtfertigung. Richtig und wichtig ist aber die Feststellung, dass jede Ernährungsweise, auch die vegane, mit Beeinträchtigungen für andere Lebewesen einhergeht und ethische Probleme aufwerfen kann.
In die ersten drei Viertel des Buches sind Erfahrungsberichte von Menschen eingestreut, die ihre Ernährung aufgrund ernster Gesundheitsprobleme – etwa Zöliakie, Diabetes oder Asthma – auf Paleo umgestellt haben. Das letzte Viertel des Buches nehmen die Kochtipps ein, die kein Paleo-Kochbuch ersetzen wollen, sondern wirklich Basics vermitteln – etwa die Herstellung von Ghee, Joghurt, Leberwurst oder Sauerkraut. Sogar Bezugsquellen sind angegeben. So, und jetzt habe ich Lust, Leberwurst zu machen! Ob ich mich traue? 🙂

Zwei neue Skizzen fürs Buch, inspiriert durch An Goris und Adrian Liston, „The immunogenetic architecture of autoimmune disease„, 2012 (Open Access):
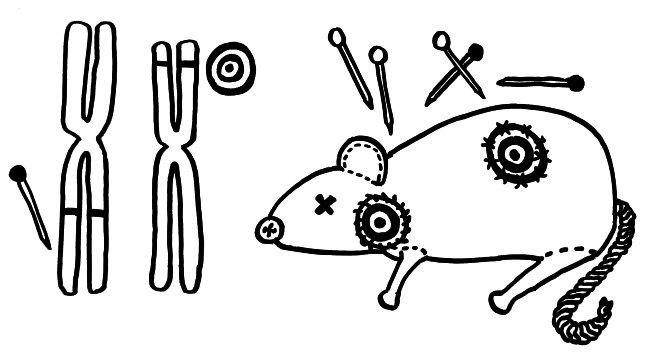
Nur wenige Autoimmunerkrankungen folgen einem einfachen Mendel’schen Erbgang. Meist sind zahlreiche Genvarianten beteiligt, die das Erkrankungsrisiko für sich genommen – wenn überhaupt – nur minimal steigern und erst gemeinsam zum Ausbruch führen. Dabei tragen einige Genvarianten zur allgemeinen Neigung des Immunsystems zu Überreaktionen bei (Voodoo-Nadeln), und andere legen fest, welches Organ betroffen sein wird (Zielscheiben).
NOD-Mäuse wurden als Typ-1-Diabetes-Modell gezüchtet; normalerweise wird ihre Bauchspeicheldrüse durch Autoimmunreaktionen zerstört (Zielscheibe auf dem Rumpf). Wenn man ihr Diabetes-Risikoallel H2g7, das zum HLA-Komplex gehört, durch die Genvariante H2h4 ersetzt, bleiben die Tiere nicht etwa gesund: Sie bekommen eine Schilddrüsen-Autoimmunerkrankung (Zielscheibe am Hals). Auch beim Menschen scheinen die meisten HLA- oder MHC-Klasse-II-Varianten auf dem 6. Chromosom festzulegen, welche Autoantigene und damit welche Organe angegriffen werden, während Risikogenorte an anderen Stellen im Genom darüber entscheiden, ob das Immunsystem überhaupt zu Autoimmunstörungen neigt.
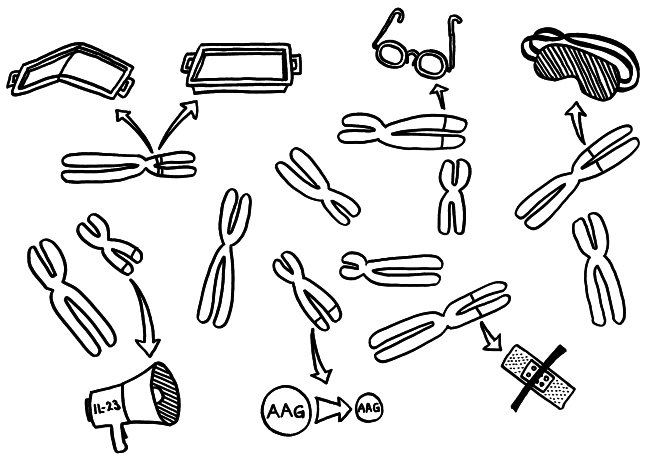
Die Genetik der Autoimmunerkrankungen ist ein etwas undankbares Forschungsfeld, auf dem man nicht hoffen darf, die eine Genvariante zu entdecken, die für einen Großteil der Erkrankungen verantwortlich ist, und daraus eine simple Therapie abzuleiten. Stattdessen kann es sein, dass jemand chronisch krank wird, weil
Auch diese Darstellung der Polygenie der Autoimmunerkrankungen ist noch stark vereinfacht – von den Wechselwirkungen zwischen unseren Genprodukten und dem Mikrobiom, unserer Nahrung, Krankheitserregern und weiteren Umweltfaktoren einmal ganz abgesehen.
Wenn also der nächste Wunderheiler um die Ecke kommt, der behauptet, man müsse nur ein bestimmtes Vitamin weglassen oder ein Mineralpräparat zu sich nehmen, um von einer nahezu beliebigen Autoimmunerkrankung geheilt zu werden: bitte auslachen.
Mustergültige, klassische Autoimmunerkrankungen erfüllen die Witebsky-Rose-Kriterien:
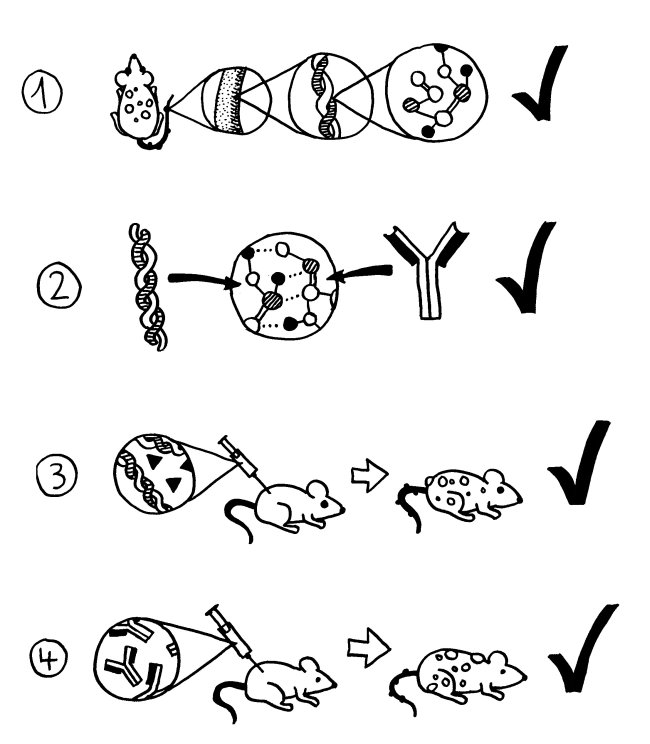
Bei etlichen Störungen, die ziemlich sicher Autoimmunerkrankungen sind, scheitern wir allerdings schon am ersten Kriterium.
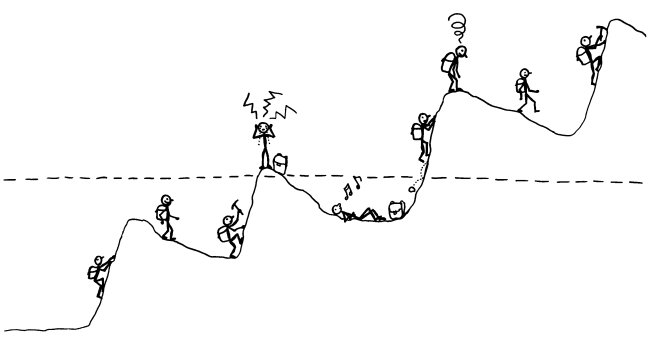 Viele Autoimmunerkrankungen verlaufen schubförmig. Am bekanntesten ist das bei der schubförmig remittierenden Multiplen Sklerose (RR-MS). Aber auch bei Typ-1-Diabetes kann auf den ersten Ausbruch von Symptomen, der zur Diagnose führt, eine Zeit der scheinbaren Genesung folgen – die sogenannte Honeymoon-Phase. Und bei den meisten Autoimmunerkrankungen geht der symptomatischen Phase (oberhalb der gestrichelten Linie) unbemerkt eine langjährige Entgleisung des Immunsystems voran, bei der nach und nach mehr Autoantikörper oder autoreaktive T-Zellen entstehen und es den regulatorischen T-Zellen immer schwerer fällt, diese selbstzerstörerischen Elemente in den Griff zu bekommen.
Viele Autoimmunerkrankungen verlaufen schubförmig. Am bekanntesten ist das bei der schubförmig remittierenden Multiplen Sklerose (RR-MS). Aber auch bei Typ-1-Diabetes kann auf den ersten Ausbruch von Symptomen, der zur Diagnose führt, eine Zeit der scheinbaren Genesung folgen – die sogenannte Honeymoon-Phase. Und bei den meisten Autoimmunerkrankungen geht der symptomatischen Phase (oberhalb der gestrichelten Linie) unbemerkt eine langjährige Entgleisung des Immunsystems voran, bei der nach und nach mehr Autoantikörper oder autoreaktive T-Zellen entstehen und es den regulatorischen T-Zellen immer schwerer fällt, diese selbstzerstörerischen Elemente in den Griff zu bekommen.

Im Jahr 2011 hatten in den Vereinigten Staaten etwa 11 Millionen Menschen Krebs und nach Schätzung der National Institutes of Health (NIH) etwa 23,5 Millionen Menschen Autoimmunerkrankungen. Die American Autoimmune Related Disease Association (AARDA) geht sogar von etwa 50 Millionen betroffenen Amerikanern aus.
Im Jahr 2003 standen in den USA zur Erforschung von Krebs gut 6 Milliarden US-Dollar zur Verfügung. Die Autoimmunerkrankungen wurden mit knapp 600 Millionen Dollar erforscht. Neuere Zahlen kenne ich nicht.