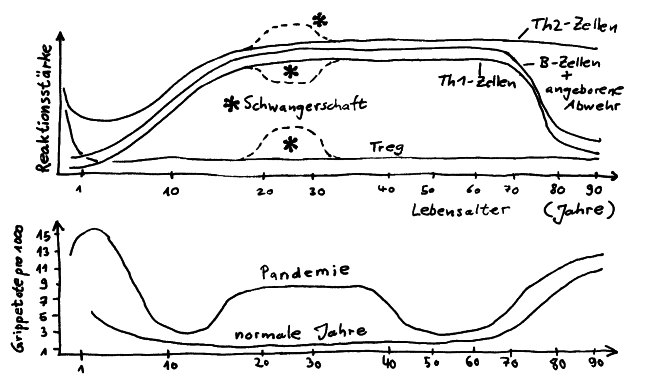
Reaktionsstärke der Hauptkomponenten des Immunsystems (oben) und Grippetote pro 1000 Personen (unten) im Lebensverlauf, nach Simon 2015
Übersichtsarbeiten, die die Entwicklung des Immunsystems von der Wiege bis zur Bahre vorstellen, sind erstaunlich selten; vermutlich ist das Thema „zu groß“. (Was soll ich da erst sagen: In meinem Manuskript ist das einer von fünf Teilen …) Im Folgenden werte ich eine 2015 erschienene Arbeit von A. Katharina Simon et al. aus: Evolution of the immune system in humans from infancy to old age.
1. Schwangerschaft und Geburt
1.1 Angeborene Abwehr
Reife neutrophile Granulozyten (kurz: Neutrophile) treten ab dem Ende des ersten Trimesters auf. Kurz vor der Geburt steigt ihre Zahl stark an, angeregt durch den Granulozyten-Kolonie-stimulierenden Faktor. Sie zeigen allerdings nur schwache Reaktionen auf Bakterien und Entzündungssignale, eine geringe Adhäsion an Endothelzellen und eine schwache Chemotaxis – insbesondere bei Frühchen.
Bei Frühchen und normalen Geburten gibt es anfangs nur wenige pulmonale Makrophagen, ihre Zahl steigt aber innerhalb weniger Tage auf Adult-Niveau an.
Neugeborene haben auch nur wenige dendritischen Zellen vom myeloiden Typ (mDCs), und diese weisen weniger HLA-Klasse-II-, CD80- und CD86-Oberflächenmarker auf als bei Erwachsenen. Daher fällt das Priming von Th1- und CD8+-T-Zellen schwächer aus, sodass Neugeborene empfindlicher für Vireninfektionen, Mycobacterium tuberculosis und Salmonellen sind als größere Kinder und Erwachsene.
Bei den plasmacytoiden dendritische Zellen (pDCs) von Neugeborenen ist die Ausschüttung von IFN-α/β nach viraler Stimulation gehemmt, was zu einer schwachen angeborenen Abwehr von respiratorischen Synzytial-Viren, Herpes simplex und Cytomegalovirus führt.
Natürliche Killerzellen (NK-Zellen) werden normalerweise durch inhibitorische Rezeptoren für HLA-A, -B, -C und -E reguliert. In der frühen Schwangerschaft reagieren sie aber kaum, wenn eine Zelle – etwa im Trophoblast – kein klassischen HLA-Klasse-I-Merkmale aufweist; außerdem lassen sie sich sehr leicht durch TGF-β supprimieren. Neonatale NK-Zellen sind weniger leicht durch IL-2 und IL-15 aktivierbar als adulte und stellen weniger IFN-γ her.
Im Serum von Neugeborenen sind fast alle Komponenten des Komplementsystems zu 10-80 % schwächer vertreten als bei Erwachsenen. Da es in Neugeborenen noch wenig Immunglobulin gibt, wird das Komplementsystem eher auf dem alternativem Weg oder auf dem Lektin-Weg aktiviert, getriggert durch Polysaccharide und Endotoxine.
Alles in allem reagiert die angeborene Abwehr bei der Geburt gedämpft. Sie muss wohl schwach ausfallen, um während der Schwangerschaft maternale Antigene und Umbaumaßnahmen zu tolerieren.
1.2 Erworbene Abwehr
Einfach positive CD4+- und CD8+-T-Zellen treten im menschlichen Thymus bereits ab Woche 15 auf und sind auch in der Peripherie schon lange vor Geburt zahlreich vertreten. Die T-Zellen funktionieren allerdings anders als später: Zur Geburt sind etwa 3% der peripheren T-Zellen Tregs (viel mehr als bei Erwachsenen); das Immunsystem hat insgesamt ein entzündungshemmendes Profil. Wird das fetale oder neonatale Immunsysteme durch fremde Antigene aktiviert, kommt es vor allem zu einer Th2-Antwort, verstärkt durch die neonatalen DCs.
Bei der Geburt sind fast alle T-Zellen naiv (d. h. noch ohne Antigen-Kontakt). In Neugeborenen treten viele T-Zellen mit γδ-T-Zell-Rezeptoren (TCRs) sowie „innate-like“ αβ-TCR-T-Zellen auf, die zwischen angeborener und erworbener Abwehr angesiedelt sind – darunter invariant natural killer T cells (iNKT), die schnell IFN produzieren, mucosal-associated invariant T cells (MAIT) und CXCL8-absondernde naive T-Zellen.
MAIT-Zellen entwickeln sich im Thymus; ihre Reifung können sie schon vor der Mikrobiom-Kolonisation in fetalen Schleimhäuten durchlaufen. CXCL-8-produzierende T-Zellen können in Neugeborenen antimikrobielle Neutrophile und γδ-T-Zellen aktivieren; sie sind vor allem in den Schleimhautbarrieren von Frühchen und normalen Neugeborenen aktiv. γδ-T-Zellen können nach schneller polyklonaler Expansion viel IFN-α herstellen und so die Unreife der klassischen Th1-Reaktion bei Neugeborenen ausgleichen.
B-Zellen: B1-Zellen schütten spontan schwach affines IgM aus, das eine eingeschränkte AG-Spezifität (gegen die gängigsten bakteriellen Polysaccharide) hat, außerdem IL-10 und TGF-β. So wird eine Th2-Antwort gefördert. Bei der Geburt sind etwa 40% der peripheren B-Zellen B1-Zellen; der Anteil der B2-Zellen nimmt später zu. [Achtung: B1/B2 sind beim Menschen noch immer nicht eindeutig belegt!]
Die meisten Antikörperreaktionen sind auf T-Zell-Hilfe angewiesen; diese wird aber durch den Mangel an Korezeptoren auf den neonatalen B-Zellen erschwert. Auch für das Komplement-Fragment C3d gibt es nur wenige Rezeptoren, sodass die Reaktion auf Polysaccharid-Komplement-Komplexe schwach ausfällt. Insgesamt ist die humorale Abwehr schwach, es gibt wenig Ig-Klassenwechsel, aber es entstehen schon Gedächtnis-B-Zellen. Bei bis zu zwei Monate alten Babys gibt es wenig somatische Hypermutation und wenig Affinitätsreifung der Antikörper. Das Knochenmark-Stroma ist noch nicht imstande, Plasmablasten lange zu unterstüzen und zu Plasmazellen reifen zu lassen; daher nimmt die Konzentration von IgG nach einer Immunisierung rasch ab. Entsprechend hoch ist die Neugeborenensterblichkeit in Populationen mit hoher Pathogenbelastung.
2. Kinder und Erwachsene
Ein wichtiger frühkindlicher Schutz gegen Infektionen, die die Mutter schon hatte, ist mütterliches IgG. Diese Antikörper werden durch die Plazenta und nach der Geburt mit der Milch übertragen. Auch 20-30 Jahre nach der Infektion der Mutter werden noch genug Antikörper übertragen, um das Kind zu schützen. Sobald das mütterliche IgG zurückgeht, sind die Kinder besonders empfindlich, da ihre eigene Antikörperproduktion noch nicht ausreicht. Heutzutage stimuliert man das kindliche Immunsystem durch Impfungen.
Während der Kindheit geht der Anteil der Tregs zurück; dafür kommen Gedächtnis-, Th1-, Th17- und Th2-Zellen hinzu, bis diese zusammen etwa so zahlreich sind wie die naiven T-Zellen. Viele der Gedächtnis-T-Zellen wurden durch das Mikrobiom geprimed, können aber später auf Pathogen-Antigene (auch aus Viren, z. B. HIV-1) kreuzreagieren, da die Antigen-Erkennungssequenzen für die T-Zell-Rezeptoren sehr kurz sind.
Ein Schutz durch die erworbene Abwehr hält nach einmaliger Infektion meist lebenslang. Gedächtnis-B-Zellen werden im Knochenmark am Leben gehalten. Teils bleiben auch die Antigene jahrelang in den Lymphknoten erhalten und werden von follikulären DCs präsentiert, die so für eine gelegentliche Teilung und Antikörper-Ausschüttung der passenden B-Zellen sorgen.
3. Weibliches Immunsystem in der Schwangerschaft
Mechanismen auf der mütterlichen Seite der Plazenta verhindern die Abstoßung des Fetus, z. B. über nicht klassische, nicht polymorphe HLA-Antigene, die örtliche Suppression durch infiltrierte NK-Zellen, Monozyten und Tregs sowie die Verhinderung der T-Zell-Aktivierung durch Tryptophan-Entzug.
Das mütterliche Immunsystem verschiebt sich während der Schwangerschaft von Th1 zu Th2 (siehe Abb.). Oft geht das mit einer Remission von Autoimmunerkrankungen einher.
4. Krebs und Autoimmunerkrankungen
Das Immunsystem bekämpft nicht nur Pathogene, sondern auch mutierte Zellen, die sich zu einem Tumor auswachsen könnten. Viele Tumoren schalten Tumor-Antigen-spezifische T-Zellen ab, indem sie an Checkpoint-Rezeptoren wie PD-1 oder CTLA4 binden. Therapien, die das verhindern, können Autoimmunerkrankungen auslösen – ebenso wie ein passiver Transfer von Anti-Krebs-T-Zellen. Überreaktionen wie Autoimmunerkrankungen oder Allergien sind der Preis, den wir für die Krebsbekämpfung durch T-Zellen zahlen.
Der Balanceakt zwischen Immunreaktionen, die Tumoren bekämpfen, und Autoimmunerkrankungen misslingt vor allem im Alter: Ein Drittel aller alten Menschen in den westlichen Ländern bekommt Krebs, 5-10% entwickeln Autoimmunerkrankungen.
Mikroorganismen wie EBV, Hepatitis B und C, HPV und Helicobacter pylori verursachen etwa ein Viertel aller Krebserkrankungen. Die chronischen Infektionen werden von spezifischen T-Zellen in Schach gehalten; im Alter kann diese Abwehr versagen kann.
5. Alter
Im hohen Alter steigt das Risiko akuter viraler und bakterieller Infektionen, außerdem ist die Sterblichkeit unter Infizierten im Alter dreimal so hoch wie bei jüngeren Erwachsenen. Bei einer normalen Grippewelle sind etwa 90% der Toten über 65 Jahre alt (s. Abb.).
Das Gleichgewicht zwischen Mikrobiom und Wirt kann durch ein nachlassendes Immunsystem gestört werden. Eine reduzierte mikrobielle Vielfalt im Darm korreliert mit Clostridium-difficile-assoziierter Diarrhö, die oft bei Alten in Krankenhäusern auftritt. Proinflammatorische Pathobionten nehmen im hohen Alter zu, immunmodulierende Arten ab.
Autoimmunerkrankungen werden im Alter häufiger, evtl. durch Lymphopenie, den Rückgang von Tregs und/oder die nachlassender Aufräumtätigkeit der Makrophagen. Der Thymus-Output sinkt, es gibt weniger neue naive T-Zellen. Auch die Fähigkeit, ein Gedächtnis für neue Antigene anzulegen, lässt nach. Das CD4+/CD8+-Verhältnis wird größer: Die Notwendigkeit, latente Viren wie EBV oder CMV zu kontrollieren, lässt weniger Platz für CD8+-Zellen. Naive B-Zellen werden zunehmend durch Gedächtnis-B-Zellen ersetzt, von denen einige “erschöpft” sind. Der Rückgang der naiven Zellen hat aber meist keine dramatischen Folgen, da alte Menschen schon über große „Gedächtnis-Datenbanken“ zu vielen Pathogenen verfügen.
Auch die angeborene Abwehr lässt im Alter nach. Die Hämatopoese verschiebt sich zugunsten myeloider Zellen – evtl. eine evolutionäre Anpassung, da zur Beseitigung der vielen seneszenten Zellen mehr Phagozytose vonnöten ist. Im hohen Alter sind Neutrophile, Makrophagen und DCs weniger leistungsfähig (weniger HLA-Expression, weniger Phagozytose …), sodass die immunologisch stille Beseitigung apoptotischer und seneszenter Zellen nicht mehr gelingt. Dann kommt es zu einer dauerhaften schwachen Entzündung (mehr proinflammatorische Zytokine: IL-1β, IL-6, IL-18 und TNF-α), die zu Atherosklerose, Demenz oder Krebs beitragen könnte.
Die zellulären und molekularen Grundlage der Immunoseneszenz sind noch nicht aufgeklärt. Ältere Zellen zeichnen sich durch drei Eigenheiten aus: (1) Telomere verkürzt -> Die Zellteilungsfähigkeit lässt nach. (2) Mitochondrien-Dysfunktion -> mehr reaktive Sauerstoffspezies. (3) Sekretion entzündungsfördernder Zytokine, Chemokine und Proteasen. Die Auswirkungen auf das Immunsystem: Mitotische Zellen wie hämatopoetische Stammzellen, T-Zellen usw. schwinden, postmitotische Immunzellen wie Neutrophile werden dysfunktional.
Hochbetagte sowie Menschen mit Autoimmunerkrankungen oder chronischen Vireninfektionen haben vor allem CD27–CD28–-T-Zellen mit sehr kurzen Telomeren, die sich kaum noch teilen können, aber noch starke Effektorfunktionen ausüben.
Bei oxidativem Stress (etwa durch reaktive Sauerstoffspezies) können DNA-Stränge zerbrechen. Verursacht wird der oxidative Stress evtl. durch ein Nachlassen der Autophagie: Altes zytoplasmatisches Material wird nicht mehr zum sicheren Abbau in Lysosomen ausgelagert.