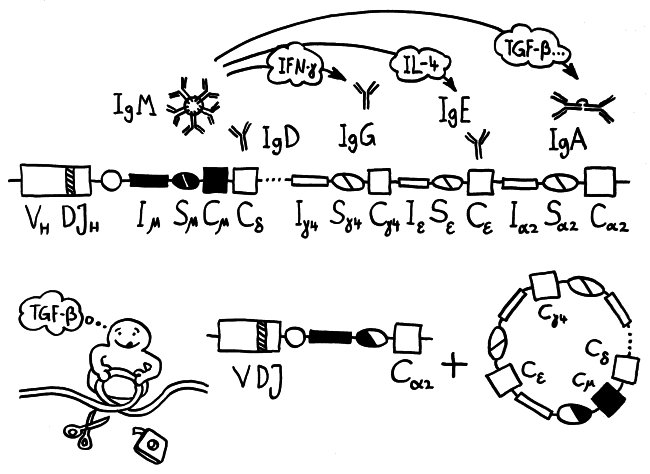
In den Follikeln des sekundären und tertiären Lymphgewebes kommt es nicht nur zum Immunglobulin-Klassenwechsel, den ich im letzten Beitrag skizziert habe, sondern auch zur Affinitätsreifung durch somatische Hypermutation und anschließende Selektion auf verbesserte Antigen-Bindungsstärke:
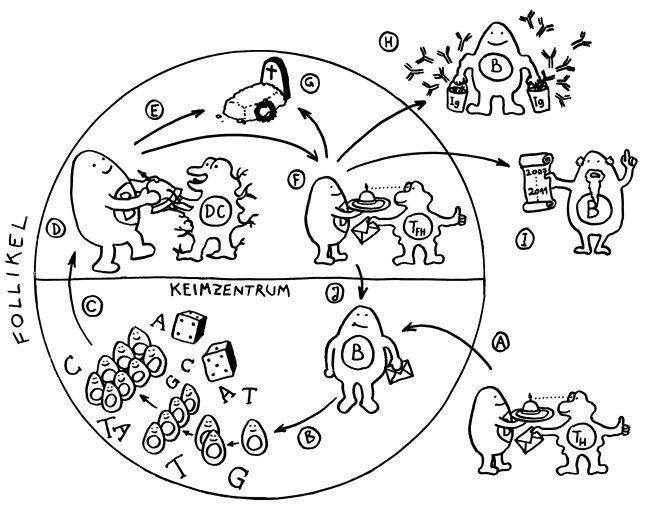
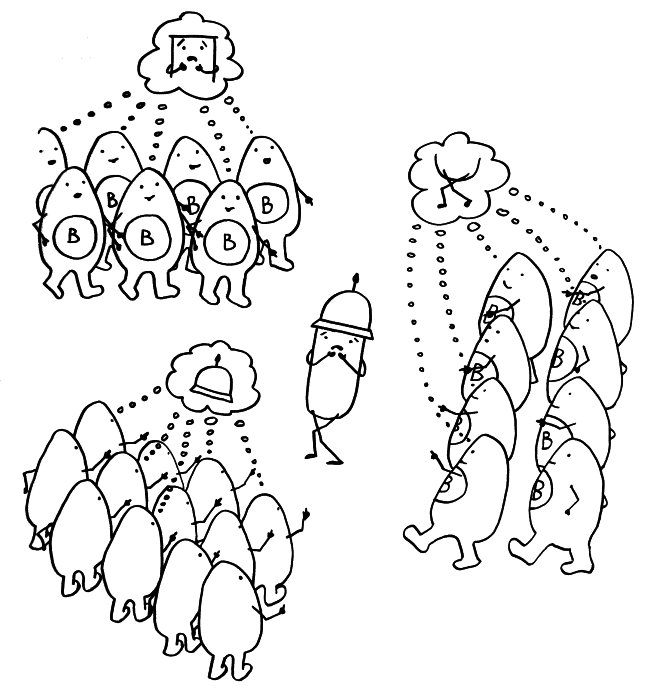
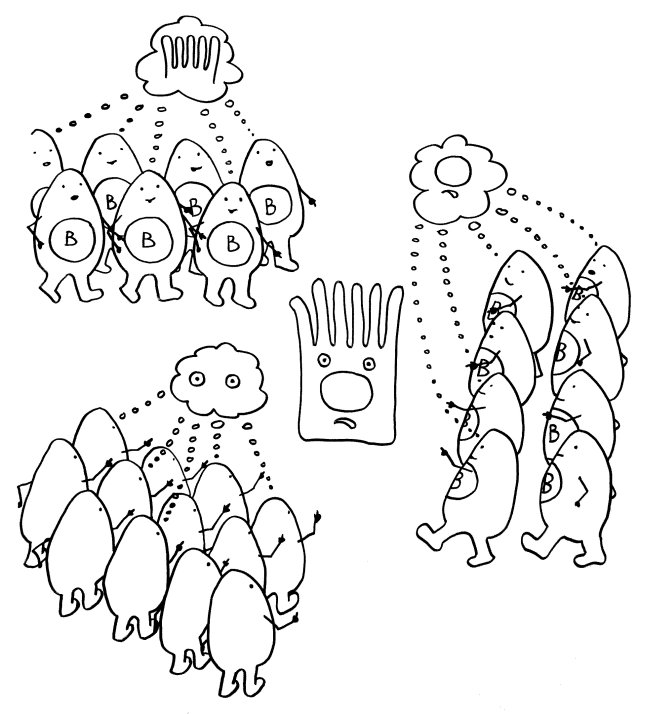
Im Uhrzeigersinn, bei 4 Uhr beginnend:
A Eine B-Zelle, die ein Antigen aufgenommen hat, präsentiert ihren Fund einer T-Helferzelle und wird vollends aktiviert, sofern der T-Zell-Rezeptor das Antigen erkennt. Sie erhält von der T-Helferzelle die Lizenz, in das Keimzentrum des Follikels einzutreten.
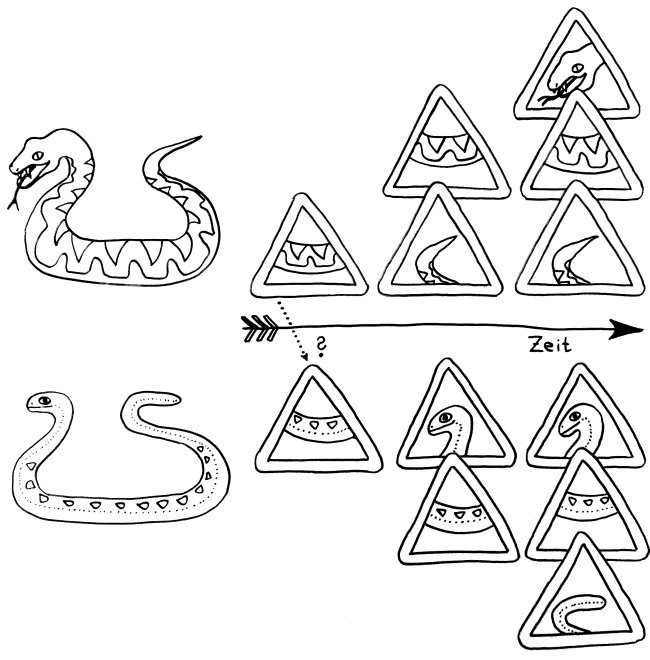
B Im Keimzentrum des Follikels vermehrt sich die B-Zelle stark durch Teilung. Währenddessen verändert das Enzym AID in dem Gen, das die antigenspezifische Bindungsstelle des Immunglobulins codiert, nach dem Zufallsprinzip einzelne Basen (A, T, C, G). Diesen Vorgang nennt man somatische Hypermutation.
C Die B-Zellen treten aus der dunklen Zone des Keimzentrums in die helle Zone über, wo sie von dendritischen Zellen (DC) erwartet werden und nach der Mutation eine Selektion durchlaufen.
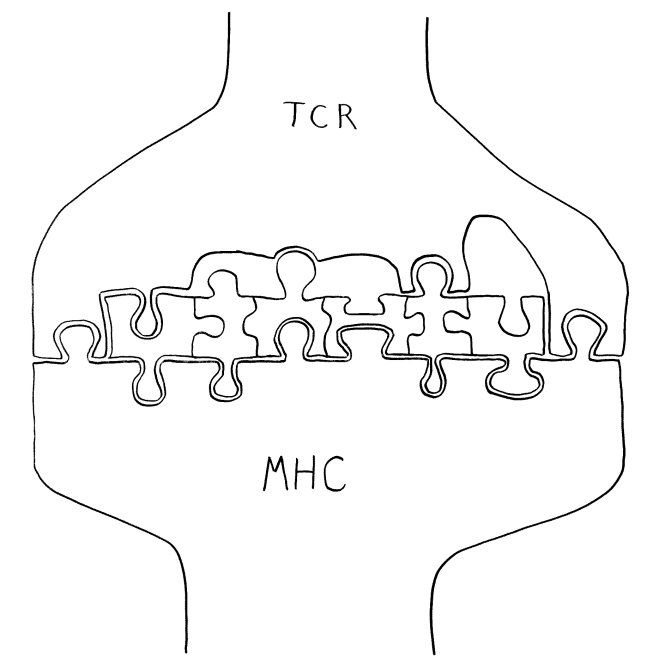
D Die dendritischen Zellen präsentieren ihnen das Antigen, um die Bindungsstärke des mutierten B-Zell-Rezeptors zu prüfen.
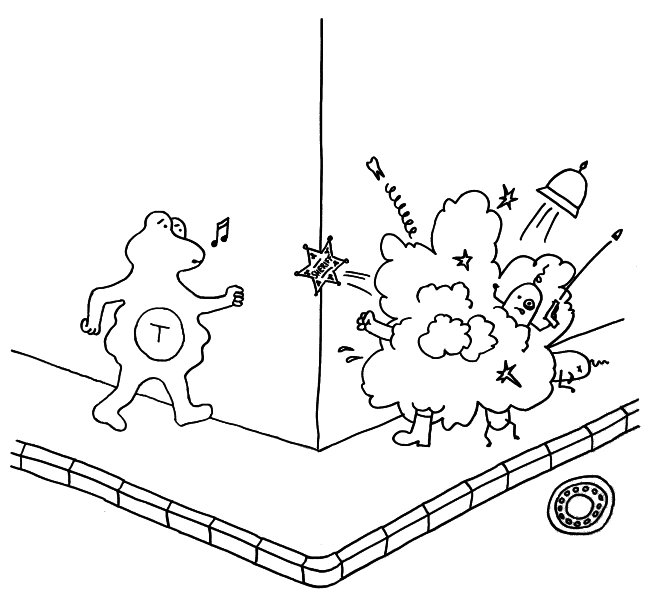
E Hat die Mutation die Bindung der Immunglobuline an das Antigen geschwächt, stirbt die B-Zelle durch Apoptose kontrolliert ab.
F Hat die Mutation die spezifische Bindung an das Antigen gestärkt, so führt die B-Zelle dieses Antigen nun auf ihrem MHC-Klasse-II-Komplex einer follikulären T-Helferzelle vor, die es mit ihrem spezifischen T-Zell-Rezeptor erkennt. Durch diesen Kontakt wird auch der Klassenwechsel bei den Immunglobulinen ausgelöst, sodass die B-Zelle nun kein IgM mehr herstellt, sondern IgG, IgE oder IgA – je nachdem, welchen Botenstoff die T-Helferzelle ausschüttet.
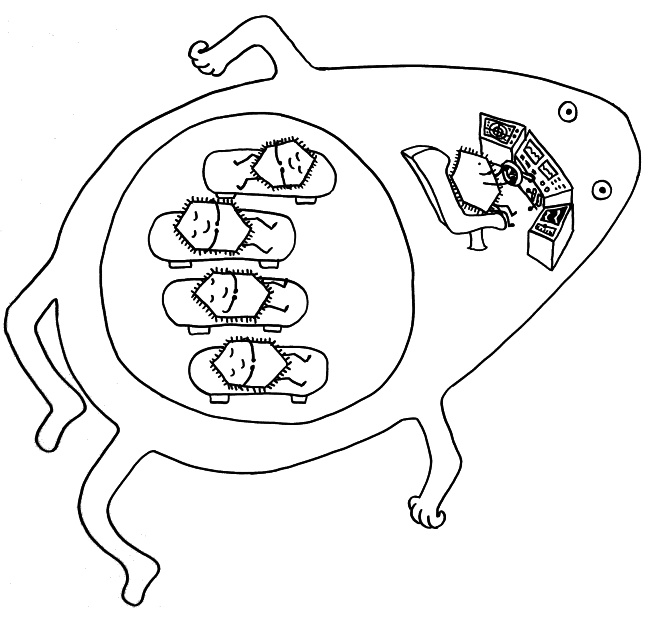
Je nach Bedarf und dem Ergebnis dieser weiteren Prüfung schlägt die B-Zelle danach einen von vier Wegen ein:
G Die B-Zelle ist unbrauchbar, weil sie der T-Zelle ihr Antigen nicht effizient präsentiert, und stirbt durch Apoptose.
H Die B-Zelle ist zur humoralen Abwehr geeignet, verlässt das Keimzentrum und entwickelt sich zur Plasmazelle weiter, die massenhaft Antikörper erzeugt.
I Einige B-Zellen reifen stattdessen zu Gedächtniszellen heran, die mit ihrem Wissen um die aktuelle Infektion dafür sorgen, dass das Immunsystem auf ein späteres erneutes Auftreten desselben Antigens schneller und stärker reagieren kann.
J Einige besonders schlagkräftige B-Zellen erhalten die Order, erneut in das Keimzentrum einzutreten, um sich zu vermehren und durch Mutation und Selektion weiter zu verbessern. So steigert der Organismus die Affinität der Immunglobuline zu einem bestimmten Antigen mit der Zeit. Diesen Vorgang nennt man Affinitätsreifung.