Zum Ausgleich für die letzten Beiträge gibt es diesmal viele Skizzen. Wie bereits erwähnt, sind die Körper weiblicher Säugetiere Mosaiken aus Zellkolonien, in denen das von der Mutter geerbte X-Chromosom stillgelegt ist, und solchen, in denen das vom Vater geerbte X-Chromosom inaktiviert wurde. Die Entscheidung fällt während der frühen Embryogenese, und zwar zufällig, und sie wird von allen Tochterzellen dieser Embryonalzellen übernommen. Nur zur Verdopplung vor einer Zellteilung werden die kompakten inaktivierten X-Chromosomen (Xi oder Barr-Körperchen genannt) kurz dekomprimiert, damit Polymerasen und andere an der Replikation beteiligte Moleküle an die DNA-Stränge herankommen. Die Zelle merkt sich aber durch epigenetische Markierungen, dass diese X-Chromosomen anschließend wieder stillgelegt werden müssen.

Unserem Körper sieht man nicht an, dass er ein solcher Flickenteppich ist. Bei Hauskatzen mit dem sogenannten Schildpatt-Muster ist das anders. Ihre Fellfarbe (rötlich oder schwarz) ist nämlich auf dem X-Chromosom codiert. Daher gibt es fast nur weibliche Schildpatt-Katzen, denn nur diese haben zwei X-Chromosomen, von denen in einigen Teilen der Haut das mütterliche und in anderen Hautpartien das väterliche Exemplar aktiv bleibt. (Die weißen – genauer: unpigmentierten – Schecken vieler Schildpatt-Katzen sind an einer anderen Stelle im Genom codiert.)
Dass die Inaktivierung eines der X-Chromosomen und damit das Schildpatt-Muster epigenetisch und nicht genetisch festgelegt ist, sieht man an der ersten geklonten Katze der Welt, die 2001 geboren wurde. Sie trägt den schönen Namen CC (für copy cat), ist genetisch mit einer Schildpatt-Katze identisch und hat dennoch ein anders gemustertes Fell.
Wie läuft die Inaktivierung ab, und wie sehr unterscheidet sich das Xi von den übrigen Chromosomen im Zellkern? Um das zu verstehen, muss man ein Missverständnis überwinden, das aus Schulzeiten stammt. Im Biologieunterricht lernt man beim Thema Zellkernteilung oder Mitose das folgende Schema auswendig, das die Phasen einer solchen Teilung zeigt. Es beginnt mit der Prophase (bei 1 Uhr) und läuft über die Metaphase (3 Uhr) und die Anaphase (7 Uhr) zur Telophase (8 Uhr). In diesen Phasen ist die Kernhülle aufgelöst, damit die Spindeln die Tochterchromatiden auseinander ziehen können. Anschließend schnürt sich die Zelle durch, und beide Tochterzellen bauen wieder Kernhüllen auf. In der Zeit zwischen zwei Zell- und Zellkernteilungen, der Interphase (12 Uhr, Sternchen), liegen die Chromosomen nicht in der uns so vertrauten, kompakten Transportform vor, sondern als dünne Schnüre, die den ganzen Kern ausfüllen.
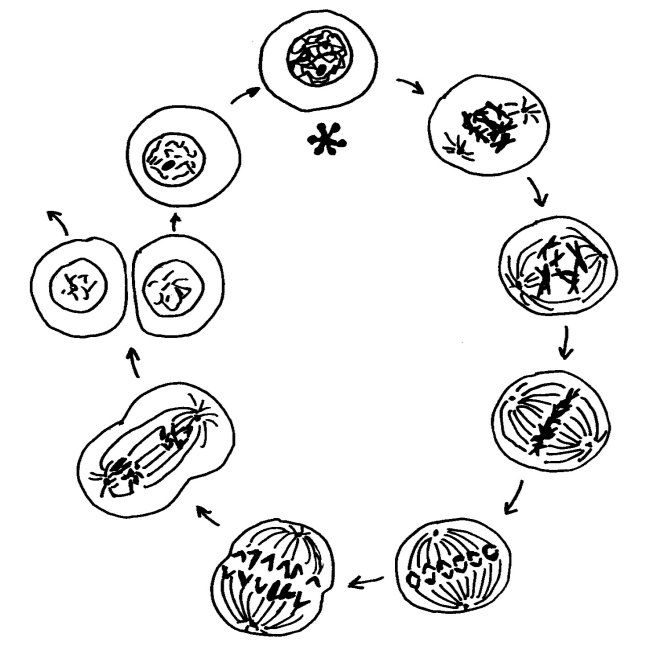
Diese Interphase ist viel, viel länger, als es das Diagramm suggeriert: Von den etwa 19,5 Stunden eines menschlichen Zellteilungszyklus entfallen etwa 18,5 Stunden auf die Interphase, in der der Zellkern wie eine Fadennudelsuppe aussieht. Nur während einer einzigen Stunde sind unsere Chromosomen so eng zusammengepackt, dass sie wie ein I oder (vor der Trennung der beiden im Zentromer zusammengehaltenen Chromatiden) wie ein X aussehen:

Der DNA-Faden wird in der Mitose um den Faktor 50.000 komprimiert, und zwar in mehreren Schritten bzw. Ebenen: von der DNA-Doppelhelix über die Perlenschnur, in der die DNA um puckförmige Proteinscheiben, sogenannte Nukleosomen, gewickelt ist, über Schlaufen erster und zweiter Ordnung bis hin zum kompletten Chromosom, das 700-mal dicker ist als eine Doppelhelix:
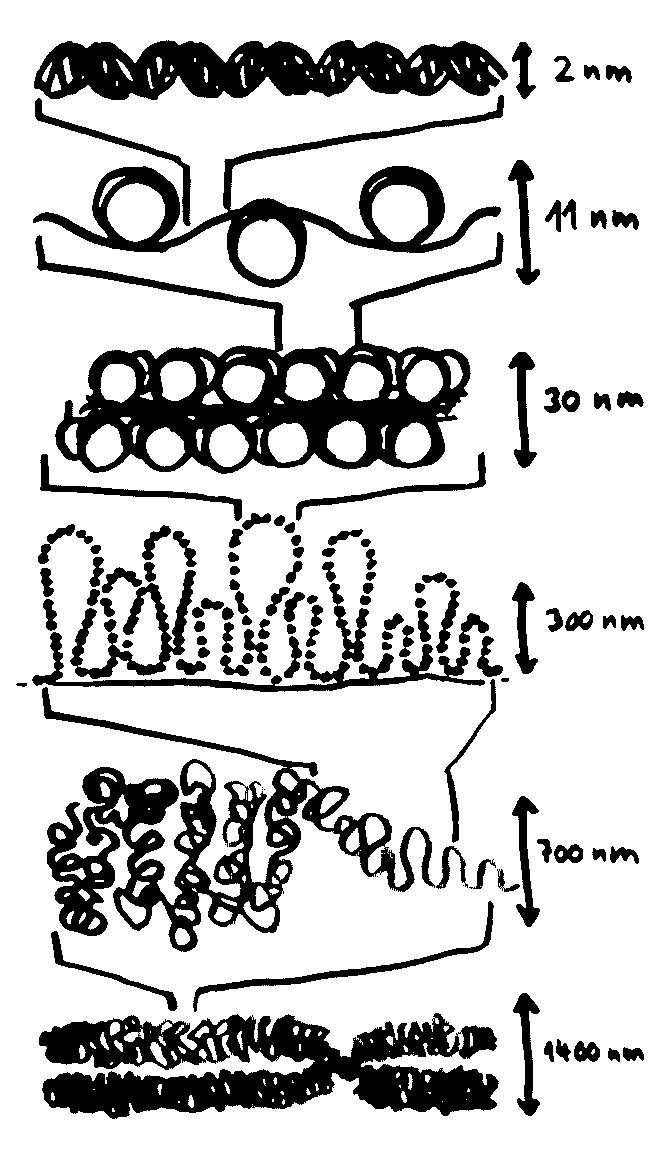
In dieser stark komprimierten Form ist das Erbgut für die Transkriptionsmaschinierie völlig unzugänglich; die Gene können also nicht abgelesen werden. In der langen Interphase sieht das anders aus: Die nun wieder lockere DNA eines jeden Chromosoms nimmt einen großen Bereich im Zellkern ein und steht an dessen Rändern mit den Nachbarchromosomen in Kontakt. Ich habe hier, damit es nicht zu unübersichtlich wird, nur ein einziges Chromosom (unten) als Faden dargestellt und von den übrigen nur die Grenzen zwischen den Regionen angedeutet: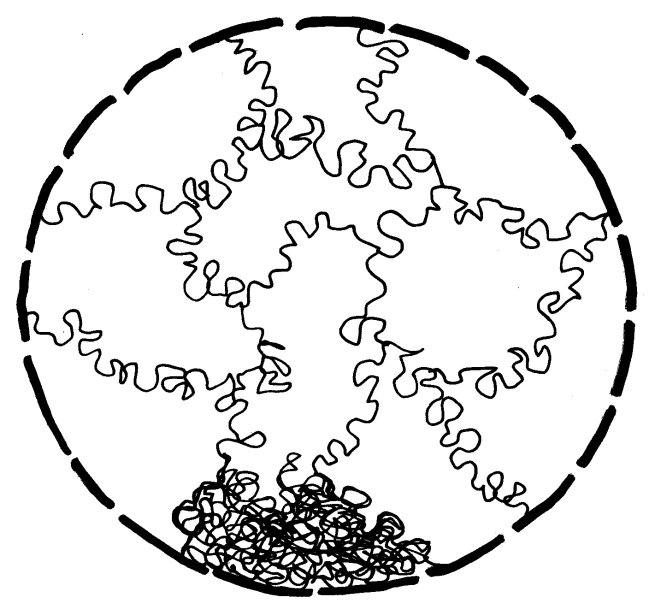
Chromosomenbereiche mit vielen Genen liegen im Allgemeinen eher in der Mitte des Zellkerns, an Genen arme Abschnitte eher in der Peripherie. Außerdem führen die Chromosomen im Kern einen komplizierten Tanz auf – wobei sie allerdings nicht so kompakt aussehen wie in dieser garantiert echten, jüngst auf einem Dachboden entdeckten Matisse-Vorstudie:
Die Chromosomen können die Position wechseln. So gelangen Gene, die gerade abgelesen werden müssen, aus der Peripherie in die Mitte, wo auch die nötigen Enzyme und Rohstoffe in höherer Konzentration vorliegen als am Rand.
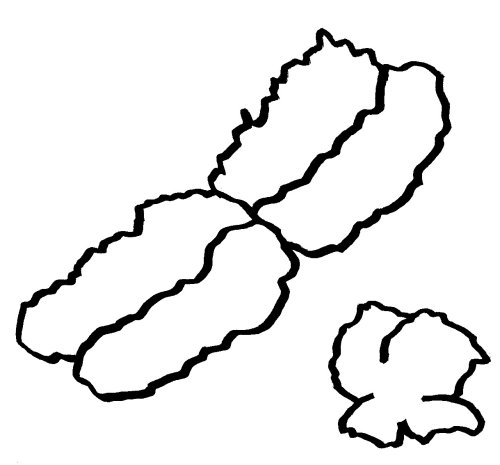
Auch die meisten unsere Geschlechtschromosomen sind die meiste Zeit keineswegs X- oder Y-förmig. Ihre Namen verdanken sie elektronenmikroskopischen Aufnahmen von Chromosomen während der Mitose. Dann sieht das bereits verdoppelte X-Chromosom mit seinem Zentromer (der Einschnürung) und seinen langen und kurzen Armen den übrigen Chromosomen recht ähnlich. Beim viel kleineren Y-Chromosom sind die Arme so kurz, dass sie kaum zu unterscheiden sind:
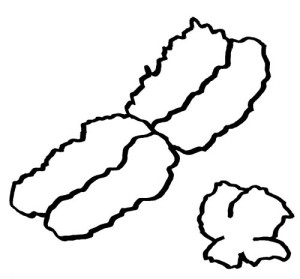
An den Enden der kurzen und der langen Arme beider Geschlechtschromosomen liegen sogenannte pseudoautosomale Regionen oder PARs. Hier sind X und Y einander so ähnlich, dass es zwischen ihnen während der Entwicklung der Keimzellen zum Austausch von Material, dem sogenannten Crossing-over kommen kann – genau wie zwischen den beiden Exemplaren eines normalen Chromosoms oder Autosoms; daher die Bezeichnung „pseudoautosomal“.
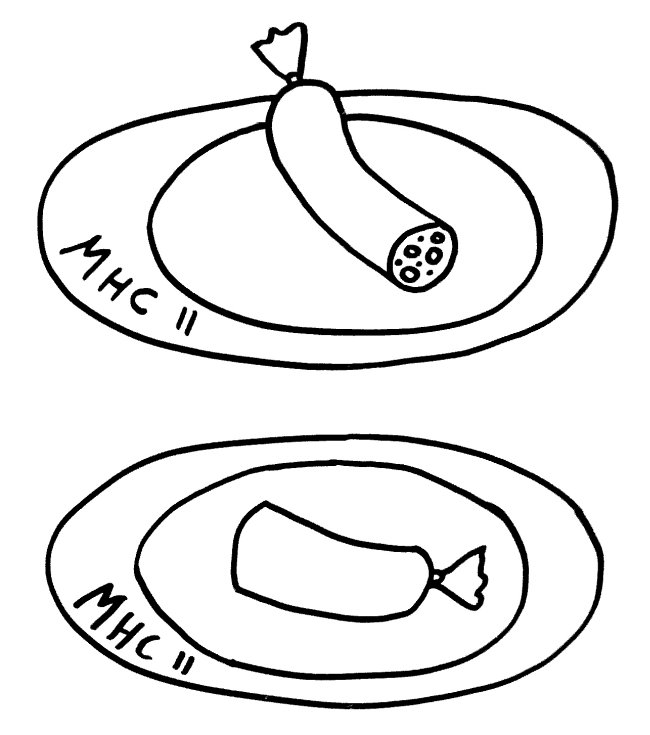
Da jede Zelle, ob männlich oder weiblich, über zwei Geschlechtschromosomen und damit über zwei PAR1 und zwei PAR2 verfügt, ist für diese Teile des X-Chromosoms keine Inaktivierung vonnöten: Ob die hier liegenden Gene nun von zwei X-Chromosomen oder von einem X- und einem Y-Chromosom abgelesen werden, ist gleichgültig. Anders sieht es mit der nicht-pseudoautosomalen X-Chromosom-Region (NPX) aus: Auf ihr liegen andere Gene als auf ihrem Pendant auf dem Y-Chromosom, MSY (für male-specific region of Y chromosome).
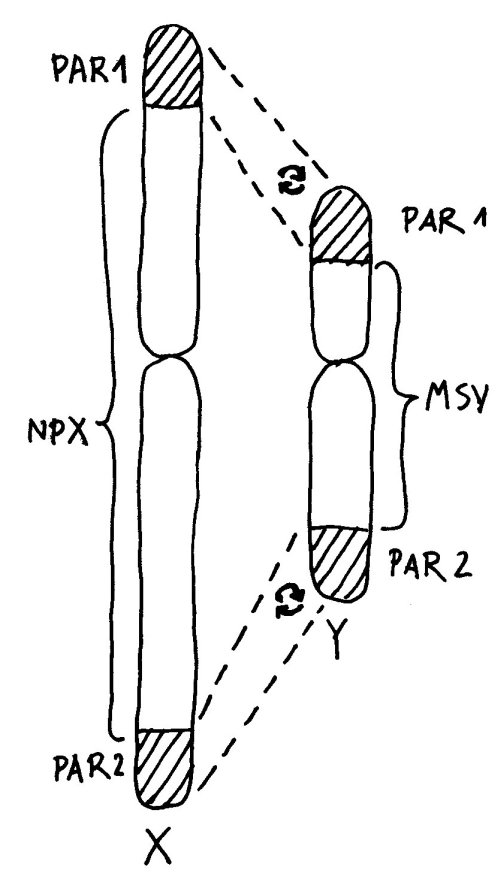
Würden diese Teile auf beiden X-Chromosomen einer weiblichen Zelle normal abgelesen, lägen ihre Genprodukte in der Zelle in doppelt so hoher Konzentration vor wie in einer männlichen Zelle. Das mag bei bestimmten Genen, die an geschlechtsspezifischen Eigenschaften mitwirken, notwendig sein. Bei vielen anderen wichtigen Genen auf dem X-Chromosom, die nichts mit dem Geschlecht zu tun haben, wäre es dagegen fatal. Daher wird ein Großteil der Gene (etwa 85 Prozent) auf einem der beiden X-Chromosomen ausgeschaltet.
Die Inaktivierung geht vom sogenannten X-Inaktivierungs-Zentrum (XIC) auf dem langen Arm des Chromosoms (Xq) aus. Dieser lange Arm wird auch als XCR (für X conserved region) bezeichnet, weil er evolutionär alte, stark konservierte Gene und Steuerungssequenzen enthält. Im Inaktivierungs-Zentrum liegt das Gen Xist, das kein Protein codiert, sondern eine RNA, die sich an alle möglichen Teile des X-Chromosoms anlagert, die sich in ihrer Nähe befinden – wobei „Nähe“ hier nicht eindimensional (benachbarte Sequenzen auf der DNA), sondern dreidimensional zu verstehen ist (im Einflussbereich liegende Schlaufen des X-Chromosom-Fadenknäuels).
Das können durchaus auch DNA-Schlaufen vom anderen Arm des X-Chromosoms sein, der jenseits des Zentromers liegt: Xp, auch XAR (für X added region) genannt, weil hier evolutionär jüngere Gene und Steuerungssequenzen liegen, die das Chromosom erst lange nach der Auseinanderentwicklung von X- und Y-Chromosom erworben hat. Auf diesem Arm liegen auch Gene, die mit einer ganzen Reihe von Autoimmunerkrankungen in Verbindung gebracht werden, etwa FoxP3 oder eine Reihe von Genen in der Region Xp22. Womöglich tragen Fehler bei der X-Chromosom-Inaktivierung zur höheren Prävalenz vieler Autoimmunerkrankungen bei Frauen bei, denn dann liegen die Produkte der fälschlich nicht inaktivierten Gene in weiblichen Zellen in stark überhöhter Konzentration vor.
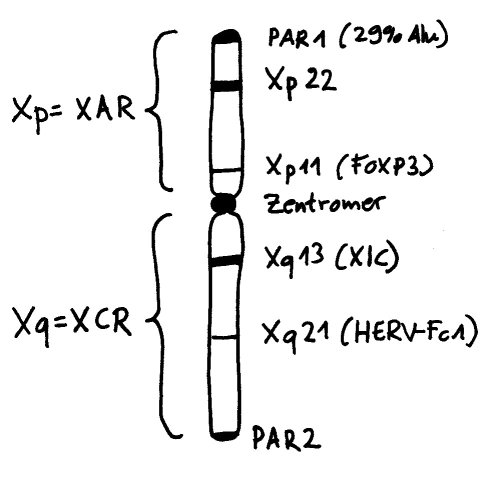
Vorlage: W. H. Brooks, Y. Renaudineau (2015), doi: 10.3389/fgene.2015.00022
Wie das X-Chromosom trotz der Barriere, die das Zentromer darstellt, so zügig inaktiviert werden kann, war der Forschung lange ein Rätsel. Studien an Mäusen helfen bei der Aufklärung (wieder einmal) nicht weiter, da deren X-Chromosom zwar ähnlich lang ist wie das der Menschen, aber kein echtes Zentromer hat, sondern eine Zentromer-ähnliche Struktur an einem Ende. Hier gibt es also keine Barriere, die von der im XIC abgelesenden RNA überwunden werden müsste:
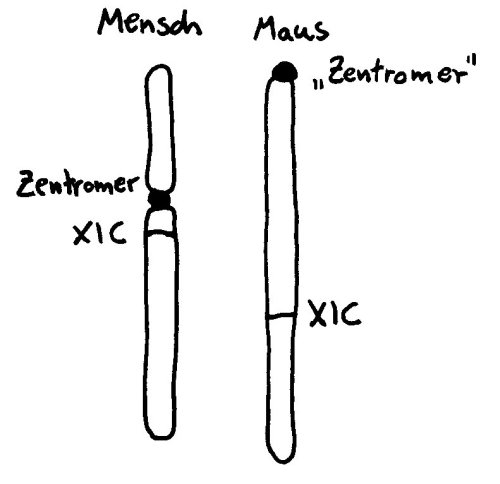
Vorlage: W. H. Brooks, Y. Renaudineau (2015), doi: 10.3389/fgene.2015.00022
Wie sich die Xist-RNA und die von ihr rekrutierten übrigen Inaktivierungsfaktoren über das X-Chromosom ausbreiten, davon hat man erst seit kurzem eine genaue Vorstellung. Demnach bilden die RNA-Moleküle (dünne geschwungene Linien mit Schlaufe) in der Umgebung des Xist-Gens (dicker schwarzer Pfeil) eine regelrechte Wolke und lagern sich an so ziemlich alle X-chromosomalen DNA-Stränge (dicke Linien) an, die in diese Wolke hineinragen. Die Histone, die die Xist-RNA nach ihrer Bindung an die DNA rekrutiert (schraffierte Scheiben und anhängende schwarze Punkte), wickeln die DNA dann eng und ordentlich zusammen:
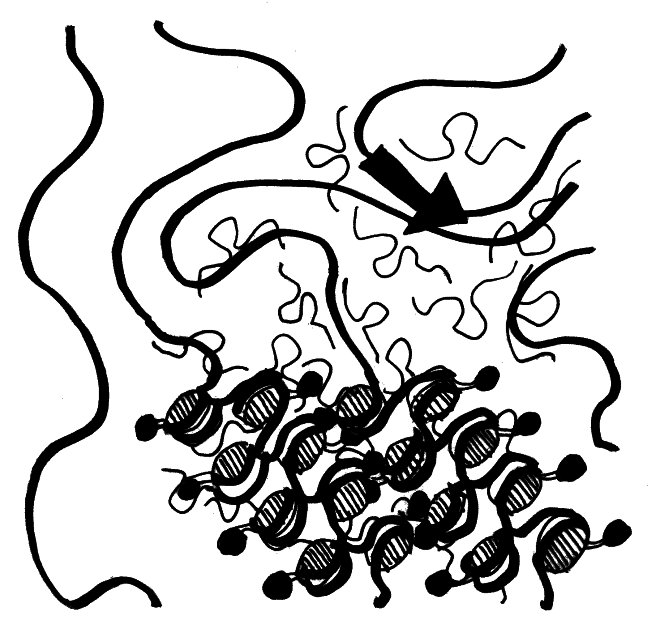
Vorlage: Engreitz et al. 2013, doi: 10.1126/science.1237973
Durch das Aufwickeln der DNA zieht der Komplex immer weitere X-chromosomale Sequenzen in die Xist-Wolke hinein. So kann sich die Inaktivierung schnell und von mehreren, nahezu beliebigen Startstellen aus über das gesamte X-Chromosom ausbreiten.
Die nunmehr stark komprimierte DNA ist nicht mehr ablesbar – genau wie in den übrigen Chromosomen während der Mitose. Allerdings ist der Komprimierungsmechanismus ein anderer als bei den Autosomen, der Komprimierungsgrad ist noch höher, und vor allem wird die Komprimierung auch während der Interphase aufrecht erhalten.
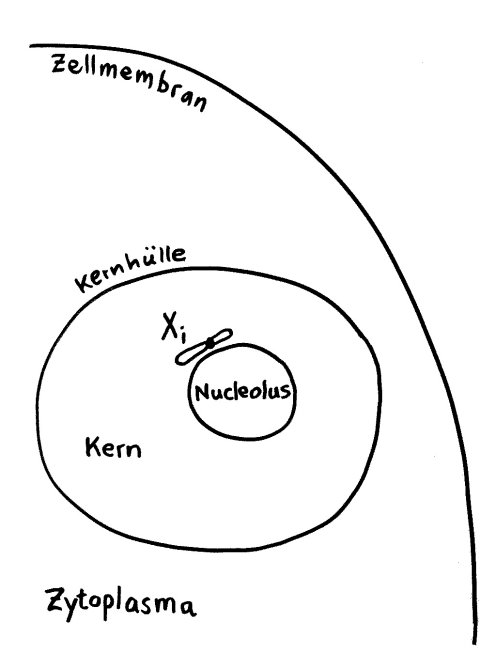
Das Xi bleibt allerdings nicht „von selbst“ inaktiv, sondern muss ständig überwacht und ggf. erneut epigenetisch markiert und verdichtet werden. Daher liegt es meist auch nicht am Rand des Kerns, sondern in unmittelbarer Nachbarschaft zum Nucleolus, einer besonders aktiven Kernregion:
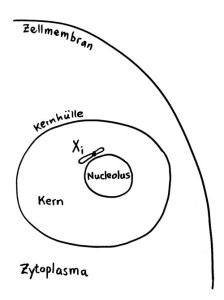
Vorlage: W. H. Brooks, Y. Renaudineau (2015), doi: 10.3389/fgene.2015.00022
Einer derzeit beliebten Hypothese zufolge können Infektionen (zum Beispiel mit Viren) Autoimmunerkrankungen auslösen, indem sie den Nucleolus anschwellen lassen: Viren kapern bekanntlich die Reproduktionsmaschinerie unserer Zellen und lassen unseren Stoffwechel in großem Stil neue Kopien ihrer selbst anfertigen. Der erhöhte Bedarf an Ribosomen und anderen Teilen der Reproduktionsmaschine führt zu einer Vergrößerung des Nucleolus. So gerät das benachbarte inaktivierte X-Chromosom in weiblichen Zellen ins Innere des Nucleolus mit seinem lebhaften Stoffwechsel:
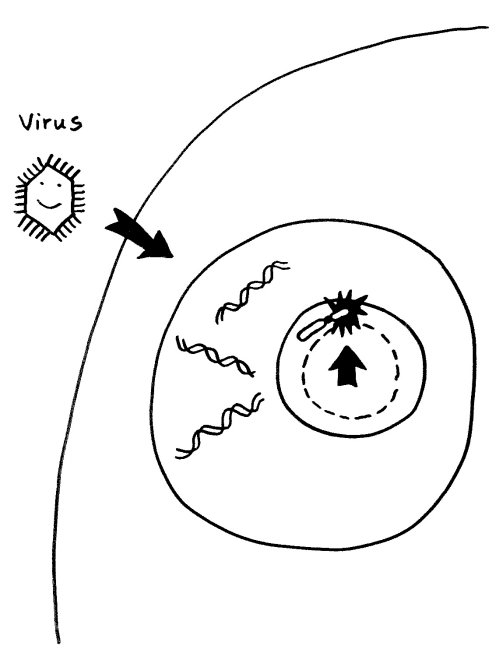
Vorlage: W. H. Brooks, Y. Renaudineau (2015), doi: 10.3389/fgene.2015.00022
Substanzen im Nucleolus, insbesondere Polyamine und RNA-Polymerase III, könnten die Inaktivierung aufheben und zum Beispiel eine massenhafte Ablesung der vielen Alu-Sequenzen im PAR1 auf dem Xp-Arm auslösen. Die so in großer Menge erzeugte Alu-RNA könnte klassische Zellkern-Proteine wie Ro und La binden und modifizieren, wodurch sich diese Proteine in Autoantigene verwandeln würden. Tatsächlich werden im Serum von Lupus-, Sjögren- oder Rheuma-Patienten häufig Anti-Ro- und Anti-La-Antikörper nachgewiesen.
Aber bislang ist das wirklich nur eine weitere Hypothese: Dass die höhere Prävalenz vieler Autoimmunerkrankungen bei Frauen auf eine unvollständige X-Inaktivierung und diese wiederum auf eine infektionsbedingte Vergrößerung des Nucleolus zurückzuführen ist, klingt plausibel, ist aber nicht belegt.
—
Literatur und Abbildungsvorlagen muss ich noch nachtragen. Jetzt: Urlaub!






















{kind=link}