Dieser Beitrag wird lang, trocken und abstrakt, und er enthält nur ein einziges eigenes Bild, und es bleibt kompliziert. Genau genommen ist er trotz langen Ringens mit dem Stoff nahezu unlesbar. Sorry. Ich musste das einfach mal notieren, um selbst nicht immer wieder durcheinander zu kommen. Im Buch landet dann eine weniger technische und überfrachtete Quintessenz.
Wann ist ein Mann ein Mann?
Und was macht eine Frau zur Frau, biologisch betrachtet? Das ist zum Glück bei Menschen, Mäusen und allen anderen Säugetieren grundsätzlich ähnlich geregelt: Neben einer Reihe „normaler“ Chromosomen, den sogenannten Autosomen, gibt es Geschlechtschromosomen – auch Gonosomen oder Heterochromosomen genannt. Während wir in jeder Körperzelle (abgesehen von unseren Keimzellen, also Eizellen oder Spermien) einen doppelten, zur Hälfte von der Mutter und zur Hälfte vom Vater stammenden Satz vom Aufbau her identischer Autosomen tragen, gilt das bei den Geschlechtschromosomen nur für die Frauen bzw. Weibchen, die von beiden Eltern je ein sogenanntes X-Chromosom erben. Männer bzw. Männchen erben dagegen von der Mutter ein X-Chromosom und vom Vater ein Y-Chromosom.
Das Y-Chromosom sieht unter dem Elektronenmikroskop im Grunde gar nicht wie ein Y aus; es ist einfach sehr klein und knubbelig und enthält viel weniger codierende, d. h. als Proteinbauanleitungen dienende Gene, nämlich 72 auf gut 57 Millionen Basenpaaren, als das X-Chromosom mit seinen 819 codierenden Genen auf 156 Millionen Basenpaaren. Evolutionär stammt das Y- wohl vom X-Chromosom ab, aber das ist schon sehr lange her: 240 bis 320 Millionen Jahre. An beiden Enden enthält es sogenannte pseudoautosomale Sequenzen, die direkte Entsprechungen auf den Enden des X-Chromosoms haben. Dadurch kommt es während der Meiose (der Reduktionsteilung, die bei der Bildung von Keimzellen den doppelten zu einem einfachen Chromosomensatz reduziert) an diesen Stellen zur Rekombination zwischen X- und Y-Chromosom, wie es sonst nur bei den Autosomen üblich ist. Die nicht pseudoautosomalen Sequenzen von X- und Y-Chromosom können dagegen schon sehr lange nicht mehr rekombinieren und tragen mittlerweile völlig unterschiedliche Gene.
Ein einzelnes Gen sorgt für Hoden
Die nicht pseudoautosomale Sequenz des kurzen Arms des Y-Chromosoms enthält das geschlechtsbestimmende Sry-Gen (für „sex-determining region of Y“), das ein Protein namens TDF codiert (für „testis determining factor“, zu Deutsch: Hoden-determinierender Faktor). Im jungen Embryo entstehen zunächst geschlechtsneutrale Keimdrüsen-Anlagen. Sobald im männlichen Embryo das Sry-Gen abgelesen wird, was beim Menschen ab der 7. Entwicklungswoche der Fall ist, regt das Protein TDF bestimmte Zellen in diesen Anlagen zur Testosteron-Produktion an, und aus den Keimdrüsen-Anlagen werden Hoden. In Abwesenheit eines Y-Chromosoms und damit des Sry-Gens entstehen dagegen Eierstöcke.
Ein Testosteron-Gen gibt es nicht
Es drängt sich die Frage auf, wo denn die Gene für Testosteron und die anderen Sexualhormone liegen – etwa auch auf den Geschlechtschromosomen? Die naive Annahme, die Bauanleitung für das männliche Sexualhormon sei wohl auf dem männlichen Geschlechtschromosom zu finden, kann nicht stimmen: Auch Frauen produzieren und benötigen Testosteron, und sie haben kein Y-Chromosom. Liegt das Gen also auf einem der Autosomen, die beiden Geschlechtern gemeinsam sind? Nein, es liegt nirgends: Es gibt kein Testosteron-Gen. Testosteron ist nämlich kein Protein, und Gene codieren nur Proteine.
Unsere Sexualhormone sind Steroidhormone, die in mehreren Schritten unter Beteiligung etlicher Enzyme aus (größtenteils vom Körper selbst hergestelltem) Cholesterin hergestellt werden, und zwar in den Keimdrüsen sowie der Nebennierenrinde. Gesteuert wird die Herstellung von der Hypophyse und dem Hypothalamus im Gehirn. Das Sexualhormonsystem ist so komplex und omnipräsent, dass es sinnlos wäre, ein bestimmtes Gen oder auch nur ein bestimmtes Chromosom für zuständig zu erklären. Einen guten Eindruck vermittelt diese Creative-Commons-Grafik:
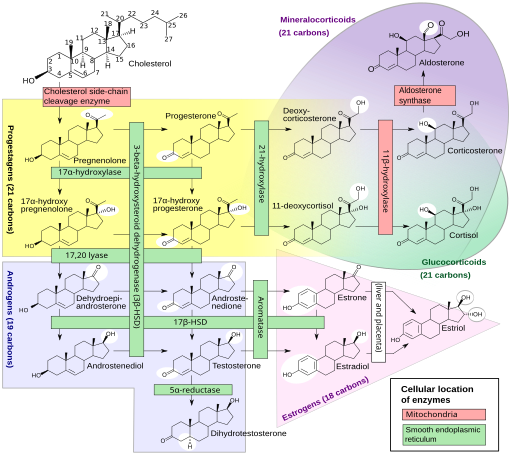
Häggström M, Richfield D (2014). „Diagram of the pathways of human steroidogenesis“. Wikiversity Journal of Medicine 1 (1). DOI:10.15347/wjm/2014.005. ISSN 20018762. (Self-made using bkchem and inkscape) [GFDL (http://www.gnu.org/copyleft/fdl.html) or CC-BY-SA-3.0 (http://creativecommons.org/licenses/by-sa/3.0/)], via Wikimedia Commons
Hormon- und Chromosomen-Wirkungen überlagern sich
An der Ausbildung der Unterschiede zwischen den Geschlechtern (beispielsweise in der Muskulatur, im Immunsystem oder im Gehirn, somit auch im Verhalten) sind weitere Gene auf dem X- und dem Y-Chromosom beteiligt, deren Wirkung nicht so gut erforscht ist wie die des Sry-Gens. Dass sie so schwer zu erforschen sind, liegt unter anderem an den Sexualhormonen, vor allem am Testosteron. Es wird bereits ab dem ersten Schwangerschaftsdrittel im Embryo produziert und prägt die gesamte weitere Entwicklung des Organismus dauerhaft. Deswegen lässt sich der reine, unverfälschte Einfluss der Geschlechtschromosomen auch nicht erforschen, indem man die neugeborenen Mäuse kastriert, sodass sie kein Testosteron mehr produzieren: Das Hormon hat dann bereits irreversible Wirkungen gezeitigt.
Selbst wenn die Hormone die geschlechtsspezifischen Effekte der Ablesung von Genen auf den Geschlechtschromosomen nicht überdecken würden, wäre es noch schwer zu ermitteln, ob und wie nun das X- oder das Y-Chromosom für einen Effekt verantwortlich ist. Wenn beispielsweise eine Erkrankung bei Männern häufiger auftritt oder dramatischer verläuft als bei Frauen, liegt das an der Ablesung eines Gens auf dem Y-Chromosom? Oder an der elternspezifischen genomischen Prägung, dem sogenannten parental imprinting, also der Tatsache, dass in allen Zellen eines Mannes das einzige, stets von der Mutter geerbte X-Chromosom aktiv ist (maternal imprinting), während im Körper einer Frau in etwa jeder zweiten Zelle das vom Vater geerbte X-Chromosom abgelesen wird (paternal imprinting)? Oder daran, dass Frauen eben zwei X-Chromosomen haben und damit u. U. über die doppelte Dosis bestimmter (in diesem Beispiel: vor Erkrankung schützender) Gene verfügen, die von der X-Inaktivierung (s. u. ) ausgenommenen sind?
Turner-Frauen, Klinefelter-Männer und der X-Dosis-Effekt
Beim Menschen lassen sich die Effekte der Geschlechtschromosomen und der Sexualhormone nicht entkoppeln, aus ethischen und aus praktischen Gründen. Gewisse Anhaltspunkte lassen sich aber aus der Untersuchung von Menschen ableiten, die nicht über den üblichen Geschlechtschromosomensatz verfügen. In der Fachliteratur werden sie häufig als experiments of nature bezeichnet.
Beim sogenannten Turner- oder Ullrich-Turner-Syndrom fehlt einer Frau eines der beiden X-Chromosomen; ihr Chromosomensatz wird als 45,X0 statt 46,XX notiert. Obwohl die Betroffenen meist ab dem Alter, in dem die Pubertät einsetzen sollte, mit Estrogen behandelt werden, hormonell also „normalen“ Frauen näherstehen als „normalen“ Männern, erkranken sie ähnlich selten wie Männer an bestimmten Autoimmunerkrankungen wie Lupus. Hier kann man einen sogenannten X-Dosis-Effekt vermuten: Irgendein Faktor, der auf dem X-Chromosom codiert ist und bei „normalen“ Frauen auf beiden Exemplaren abgelesen wird, erhöht das Erkrankungsrisiko. Hormonelle Einflüsse sind aber nicht völlig auszuschließen.
Ähnlich sieht es bei Männern aus, die neben ihrem Y-Chromosom zwei X-Chromosomen haben (Chromosomensatz 47,XXY statt 46,XY). Männer mit diesem sogenannten Klinefelter-Syndrom haben ein ebenso hohes Risiko, an Lupus zu erkranken, wie Frauen. Auch das spricht – bei aller Vorsicht wegen der gleichzeitigen Wirkung der Sexualhormone – für einen X-Dosis-Effekt.
Unvollständige X-Inaktivierung
Allerdings ist dieser X-Dosis-Effekt selbst erklärungsbedürftig. Eigentlich besagt das Dogma der X-Inaktivierung: Um die Dosis der vielen nicht für die Geschlechtsentwicklung zuständigen X-chromosomalen Gene zwischen Mann und Frau anzugleichen, wird zu einem frühen Zeitpunkt der Embryonalentwicklung in allen Zellen weiblicher Embryonen eines der beiden X-Chromosomen – und zwar zufällig das von der Mutter oder das vom Vater geerbte Exemplar – inaktiviert, indem es zu einem sehr dichten, nicht mehr ablesbaren Klümpchen verschnürt wird, dem sogenannten Barr-Körperchen. Diese Inaktivierung ist epigenetisch codiert und wird an alle durch die weiteren Zellteilungen entstehenden Tochterzellen weitergegeben. Der Körper einer Frau ist folglich ein Mosaik aus Zellkolonien, in denen das mütterliche, und solchen, in denen das väterliche X-Chromosom ablesbar bleibt.
Aber so einfach ist es nicht. Erstens scheint bei größeren X-Chromosomen-Defekten (etwa fehlenden oder verdoppelten Abschnitten) bevorzugt, d. h. nicht-zufällig, das defekte Exemplar stillgelegt zu werden. Und zweitens gibt es auf dem X-Chromosom eine ganze Reihe von Genen, die sehr wohl von beiden Exemplaren abgelesen werden. Aus dem kompakten Barr-Körperchen ragen Schlaufen nur locker aufgewickelter, sogenannter dekondensierter DNA heraus, an die die Transkriptionsmaschinerie andocken kann. Diese Stellen sind gute Kandidaten für Gene, deren Ablesung Geschlechtsunterschiede bewirkt.
Dann also Mäuse und Ratten
Wie im letzten Artikel erwähnt, lassen sich an Nagetieren (bei allen Unterschieden im Detail) Grundsatzfragen erforschen, die am Menschen niemals zu klären wären – aus ethischen und praktischen Gründen. Zufällig ist das Mäusegenom ähnlich groß wie das Humangenom: Beide umfassen etwa 23.000 bis 24.000 Gene, verteilt auf etwa drei Milliarden Basenpaare. Während Menschen 22 Autosomen-Paare sowie zwei Geschlechtschromosomen haben, verteilt sich das Hausmaus-Genom auf 19 Autosomen-Paare und ein Geschlechtschromosomen-Paar. Laborratten haben neben ihren Geschlechtschromosomen 20 Autosomen-Paare. Die Abstammungslinien der Ratten und der Mäuse haben sich vor 12 bis 24 Millionen Jahren getrennt.
Die einfachsten Experimente kommen ohne genetische Manipulation aus und wurden zum Teil schon vor einem halben Jahrhundert angestellt. Sie machen sich den aus unserer Sicht ungewöhnlichen Aufbau der Mäuse-Gebärmutter zunutze.
Testosteron: Der Positionseffekt
Die Gebärmutter der Maus besteht aus zwei Trakten, sogenannten Hörnern, die sich von den Eileitern bis zur Vagina erstrecken. Die Embryonen (bis zu einem Dutzend) liegen mit jeweils eigener Plazenta in separaten Amnionsäcken aufgereiht, sodass sie bis auf die Endpositionen je zwei Nachbarn haben. Das Testosteron, das die männlichen Embryonen von einem frühen Zeitpunkt an produzieren, diffundiert durch die Gebärmutter auch zu den nächsten Nachbarn hinüber.
Aus dieser Anordnung ergeben sich verschiedene vorgeburtliche Hormonmilieus: Sowohl männliche als auch weibliche Mäusebabies können 0 bis 2 männliche Nachbarn haben und sind entsprechend keinem (0M), wenig (1M) oder viel (2M) geschwisterlichem Testosteron ausgesetzt. Im Blut weiblicher 2M-Embryonen lässt sich deutlich mehr Testosteron und weniger Estrogen nachweisen als in weiblichen 0M-Embryonen.
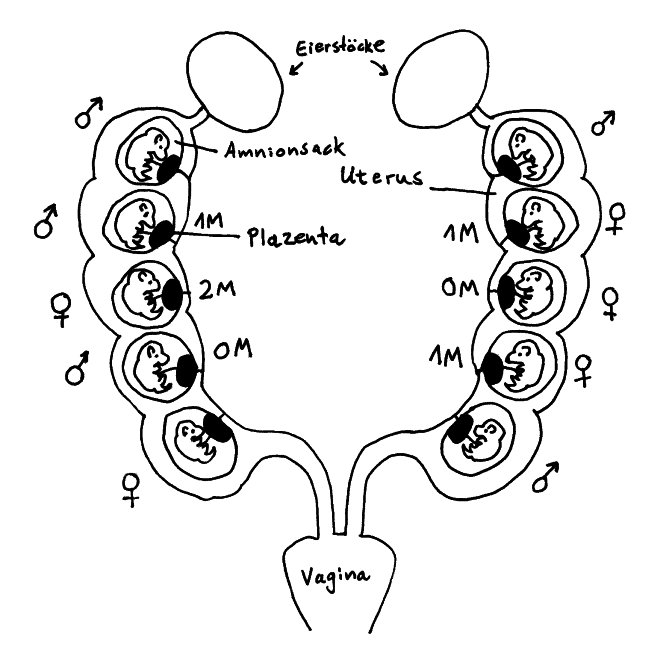
Weibliche 2M-Embryonen werden zu leicht vermännlichten Weibchen: Ihr Anogenitalabstand (der Abstand zwischen Anus und Genitalöffnung) ist größer als bei den anderen Weibchen, sie sind weniger ängstlich, verteidigen größere Reviere, werden später geschlechtsreif, wirken auf die Männchen weniger attraktiv, haben – unter anderem wegen eines längeren Zyklus – weniger Würfe, sind aber durchaus fruchtbar und verteidigen ihre Jungen energischer gegen Angriffe als die weiblicheren Weibchen. Außerdem scheinen 0M-Weibchen mehr Weibchen und 2M-Weibchen mehr Männchen in die Welt zu setzen.
Es ist anzunehmen, dass auch die genaue Zusammensetzung, die Aktivierungsschwellen und die Reaktionsstärken des Immunsystems bei 2M-Weibchen anders ausfallen als bei 0M-Weibchen, aber das scheint an Mäusen oder Ratten noch nicht näher untersucht worden zu sein. Allerdings belegten andere Versuche, bei denen man neugeborenen Ratten Testosteron in unterschiedlicher Dosis verabreicht hat, eine Vermännlichung des Immunsystems: Die Zahl der CD4+-T-Zellen im Blut sank, und die Zahl der CD8+-T-Zellen sowie der regulatorischen T-Zellen stieg an. (Auch unter uns Menschen ist der CD4/CD8-Quotient, das Zahlenverhältnis der CD4+-T-Zellen zu den CD8+-T-Zellen, bei Männern ein wenig kleiner als bei Frauen.)
Stress wirkt ähnlich
Setzt man trächtige Mäuse hellem Licht oder Hitze oder sozialem Stress durch hohe Besatzdichten der Käfige aus, so hat das auf ihre Jungen ähnliche Auswirkungen wie das Testosteron benachbarter männlicher Embryonen: Ihre Töchter haben einen größeren Anogenitalabstand und – wie die gestressten Mütter selbst – mehr Testosteron im Blut als sonst.
Einer beliebten evolutionsbiologischen Hypothese zufolge ist das kein Zufall, sondern eine Anpassung an schwankende Umweltbedingungen: In bereits dicht besiedelten, mithin stressigen und ressourcenarmen Gebieten geborene Weibchen sind auf diese Weise aggressiver, neigen zur Abwanderung, haben weniger Würfe und bekommen mehr männliche als weibliche Junge, was die Überlebenschancen des Nachwuchses steigern dürfte. In weniger dicht besiedelten Lebensräumen, also unter weniger stressigen Bedingungen ausgetragene Weibchen entsprechen eher dem 0M-Typ, sind für die Männchen attraktiver, bekommen früher und mehr Junge, unter denen überproportional viele weitere „weibliche Weibchen“ sind, die ungern abwandern. So wächst die Population schnell an.
Insgesamt scheint das System der 0M-, 1M- und 2M-Babies die Flexibilität der Tiere bei der Anpassung an unterschiedliche Umwelten und Selektionsdrücke zu gewährleisten. Da mit der örtlichen Populationsdichte auch der Pathogendruck steigt, also mehr und andere Infektionen drohen als in dünn besiedeltem Gebiet, ist anzunehmen, dass diese Variabilität auch das Immunsystem umfasst.
Genetisch veränderte Mäuse: vier Kern-Genotypen
Aber welchen hormonunabhängigen Einfluss nehmen unsere Geschlechtschromosomen auf die Ausprägung der sekundärer und tertiärer Geschlechtsmerkmale, und wie führt das zu Geschlechtsunterschieden bei Erkrankungsrisiken? Um die Wirkung der Hormone von der Wirkung X- und Y-chromosomaler Gene außerhalb der Keimdrüsen zu trennen, muss man in das Genom der Mäuse eingreifen.
Dazu kann man zum Beispiel auf eine spontane Mutation zurückgreifen, die das Sry-Gen (und wohl nur dieses) aus dem männlichen Geschlechtschromosom der Maus entfernt. Dieser Geschlechtschromosomensatz wird als XY– bezeichnet. In einem zweiten Schritt fügt man bei einem Teil der Tiere ein Sry-Gen auf einem der Autosomen, also der Nicht-Geschlechtschromosomen ein. Das Y-Chromosom ist nun nicht mehr für die Entwicklung der Keimdrüsenanlagen zu Hoden zuständig. Man erhält vier Genotypen:
- männlicher Geschlechtschromosomensatz und männliche Keimdrüsen (XYM),
- männlicher Geschlechtschromosomensatz und weibliche Keimdrüsen (XYF = XY–),
- weiblicher Geschlechtschromosomensatz und männliche Keimdrüsen (XXM),
- weiblicher Geschlechtschromosomensatz und weibliche Keimdrüsen (XXF).
Daher werden diese Mäuse als FCG mice (für four core genotypes) bezeichnet. In den meisten Versuchen kastriert man die Tiere, sobald sie geschlechtsreif sind; d. h. man entfernt die Keimdrüsen, um akute Auswirkungen der Sexualhormone auszuschließen, insbesondere die Beeinflussung der Ablesung aller möglicher Gene. Damit verringern sich die Unterschiede zwischen den Geschlechtern, aber sie verschwinden nicht, da die Keimdrüsen und ihre Hormone die Organe und Gewebe der Tiere bis zum Zeitpunkt der Kastration irreversibel geprägt haben.
Der Vergleich der beiden F-Typen mit den beiden M-Typen, also der Mäuse ohne und mit Sry-Gen, offenbart die Auswirkungen der in den Keimdrüsen produzierten Sexualhormone – u. U. vermischt mit weiteren, nicht über die Hormone vermittelten Auswirkungen des Sry-Gens. Der Vergleich der beiden XX-Typen mit den beiden XY-Typen liefert dagegen Informationen über geschlechtschromosomale Effekte.
Dabei zeigt sich zum Beispiel, dass bei bestimmten Mäusestämmen mit einer angeborenen Neigung zu den Autoimmunerkrankungen XX-Tiere mit höherer Wahrscheinlichkeit tatsächlich erkranken als XY-Tiere, und zwar unabhängig davon, ob sie mit männlichen oder mit weiblichen Keimdrüsen zur Welt gekommen sind.
Geschlechtschromosomen-Effekt beim Mausmodell für Multiple Sklerose
Ein Forscherteam um Rhonda R. Voskuhl versucht seit über einem Jahrzehnt, anhand von FCG-Mäusen zu ergründen, ob es an den Sexualhormonen oder an den sonstigen Wirkungen der Geschlechtschromosomen liegt, dass einerseits so viel mehr Frauen als Männer Autoimmunerkrankungen wie Multiple Sklerose (MS) oder Lupus bekommen, die Krankheiten andererseits bei männlichen Patienten oft dramatischer verlaufen als bei Frauen. Bereits 2005 stellten sie fest, dass bei einem Mausmodell für MS, nämlich der experimentellen Autoimmun-Enzephalitis (EAE), das männliche Sexualhormon Testosteron die Autoimmunreaktion auf das verabreichte Autoantigen hemmt: Es verlangsamt die Vermehrung autoreaktiver Immunzellen und senkt in den Lymphknoten die Konzentration der Botenstoffe TNFα, IFNΥ und IL-10. Bei kastrierten Männchen und Weibchen fiel der Unterschied geringer aus, aber er blieb erhalten. Das konnte entweder ein langfristiger Testosteroneffekt sein, etwa eine entzündungshemmende Prägung des Immunsystems oder des von der Erkrankung betroffenen Gewebes, oder ein hormonunabhängiger Geschlechtschromosomen-Effekt.
Um das zu klären, kreuzte das Team die oben eingeführten vier Genotypen in ein Mausmodell für MS ein. Bei Mäusen mit Keimdrüsen konnten sie keinen Einfluss der Geschlechtschromosomen auf die Stärke der Autoimmunreaktion feststellen, da die akute Hormonwirkung alles überdeckte. Bei Mäusen mit weiblichen Keimdrüsen, die eine Woche vor der Verabreichung des Autoantigens kastriert wurden, tat sich ein Unterschied auf: „Normale“ Weibchen (XXF) reagierten schwächer auf das Autoantigen als Weibchen mit Y–-Chromosom (XYF = XY–). In den Tieren mit einem Y–-Chromosom vermehrten sich die Immunzellen nach der Stimulation durch das Autoantigen stärker, und sie hatten mehr TNFα, IFNΥ und IL-10 im Blut. Bei kastrierten XXM- und XYM-Mäusen war es genauso, allerdings nur, wenn zwischen Kastration und Autoantigen-Gabe genug Zeit verstrich, um das restliche Testosteron im Körper abzubauen, das andernfalls den Effekt überdeckte.
Damit war klar: Die schwächere Autoimmunreaktion der Männchen im ersten Teil des Experiments war auf die langfristigen Nachwirkungen des Testosterons zurückzuführen und nicht auf eine Schutzwirkung der Geschlechtschromosomen, denn diese wirken genau umgekehrt: Ein Y-Chromosom verstärkt die Autoimmunreaktionen – oder zwei X-Chromosomen schwächen sie ab. Die gegenläufige Wirkung von Testosteron und Geschlechtschromosomen in Männchen tauften die Autoren „Yin-Yang effect“. Nun ja.
Unklar blieb zunächst, welches der drei oben genannten Szenarien hinter der stärkeren, zur beschleunigten Neurodegeneration führenden Immunreaktion bei chromosomal männlichen Mäusen mit experimenteller Autoimmun-Enzephalomyelitis steckt: bestimmte Gene auf dem Y-Chromosom, die höhere X-Chromosomen-Dosis in den XX-Mäusen oder die genomische Prägung (parental imprinting).
Ein X-Dosis-Effekt in den Immunzellen
Weitere, ähnlich angelegte Versuche desselben Teams am selben Mäusestamm erbrachten verwirrende Ergebnisse: Die Autoimmun-Enzephalitis verlief bei kastrierten XX-Tieren (mit oder ohne Sry) nicht etwa leichter, sondern klinisch schwerer als bei kastrierten XY–-Tieren. Das lag offenbar nicht an einer größeren Empfindlichkeit ihres zentralen Nervensystems für Immunreaktionen, sondern an den Autoantigen-stimulierten Immunzellen, die, wenn sie aus chromosomal weiblichen Tieren stammten, mehr Schaden anrichteten als bei einer Herkunft aus chromosomalen Männchen.
Dasselbe galt für ein anderes Mausmodell, bei dem die Tiere eine Lupus-ähnliche Autoimmunkrankheit bekommen, wenn man ihnen die Chemikalie Pristan injiziert: Tiere mit einem XX-Chromosomensatz hatten schlechtere Überlebenschancen als solche mit XY-Chromosomensatz. Bei diesen Versuchen wirkten Ying (Testosteron) und Yang (XY-Chromosomensatz) in den Männchen also nicht gegenläufig, sondern in dieselbe Richtung.
Woran lag’s? In kastrierten XY–-Tieren waren die Konzentrationen der an Th2-Immunreaktionen beteiligten Zytokine IL-5, IL-10 und IL-13 sowie in geringerem Ausmaß auch die Konzentrationen der Th1-Zytokine TNFα und IFNΥ höher als in kastrierten XX-Tieren. Zugleich exprimieren viele Immunzellen (vor allem Makrophagen und dendritischen Zellen) in XY–-Mäuse das auf dem X-Chromosom angesiedelte Gen IL-13Rα2 schwächer als XX-Mäuse. Das Gen codiert einen Interleukin-Rezeptor, der als sogenannter decoy receptor Th2-Zytokine „ködern“ oder einfangen und damit unwirksam machen kann.
Den Zellen der XX-Mäuse stehen wegen der stärkeren IL-13Rα2-Ablesung also weniger Th2-Zytokine zur Verfügung, die Autoimmunreaktionen eindämmen können und damit vor experimenteller Autoimmun-Enzephalitis schützen. Auch bei anderen Mausmodellen für entzündlichen Erkrankungen mit Immunsystem-Überreaktion wurde eine selektive Erhöhung der IL-13Rα2-Expression nachgewiesen. Am einfachsten lässt sich die höhere Dosis des (zumindest in diesem Kontext) schädlichen Köder-Rezeptors mit einer unvollständigen X-Chromosom-Inaktivierung in XX-Mäusen erklären. Demnach handelt es sich um einen X-Dosis-Effekt.
Chimären
Es ist bekannt, dass im Gehirn besonders viele der Gene auf den Geschlechtschromosomen, insbesondere auf dem X-Chromosom, exprimiert werden. Daher wollte das Forscherteam in weiteren Versuchen klären, ob und wie die Geschlechtschromosomen neben der Stärke der Autoimmunreaktionen auch die Empfindlichkeit des zentralen Nervensystems für Beschädigungen durch diese Immunreaktionen beeinflussen.
Diese Frage klärten sie mit einem weiteren Mausmodell: Knochenmark-Chimären. Durch Bestrahlung zerstörten sie das Knochenmark und damit das Immunsystem von XX- und XY–-Mäusen, die beide mangels Sry-Gen weibliche Keimdrüsen und entsprechend ein hormonell weiblich geprägtes zentrales Nervensystem hatten. Dann ersetzten sie es durch Knochenmark (und somit Immunsystem-Stammzellen) aus XX- oder XY–-Mäusen. So entstanden vier Kombinationen:
- XX-Immunsystem mit XX-Gehirn,
- XX-Immunsystem mit XY–-Gehirn,
- XY–-Immunsystem mit XX-Gehirn und
- XY–-Immunsystem mit XY–-Gehirn.
Ein Parental-Imprinting-Effekt im zentralen Nervensystem
Durch die Verabreichung von Antigenen wurde das Immunsystem dieser Chimären zu Autoimmunreaktionen im zentralen Nervensystem angeregt. Die Autoimmun-Enzephalitis verlief im XY–-Gehirn dramatischer als im XX-Gehirn: Die Tiere konnten sich bald schlechter bewegen, ihre Nervenzellen hatten weniger intakte Myelinscheiden und Axone, und im Kortex waren mehr Synapsen verloren gegangen.
In den Kortex-Neuronen von XY–-Gehirnen wurde auch der Rezeptor Tlr7 stärker exprimiert, der eine solche Neurodegeneration fördert. Die Geschlechtschromosomen-Ausstattung des Immunsystems dieser Mäuse beeinflusste den Effekt nicht. Das Gen Tlr7 liegt auf dem X-Chromosom; am Y-Chromosom kann es also nicht liegen. Ein X-Dosis-Effekt kann es auch nicht sein, denn dann müsste das Gen in XX-Mäusen stärker exprimiert werden.
Aber ein X-Chromosomen-Gen, das je nach seiner Herkunft (Mutter oder Vater) und seiner entsprechenden epigenetischen Markierung, also dem parental imprinting, unterschiedlich stark inaktiviert wird, kann den Effekt erklären. Alle X-Chromosomen in den XY–-Mäusen stammen von der Mutter, da das Y-Chromosom nur vom Vater kommen kann. Aber nur etwa Hälfte aller Zellen in XX-Mäusen exprimiert das mütterliche Tlr7-Gen; in der anderen Hälfte stammt das Gen vom Vater. Wenn die epigenetische Markierung auf dem väterlichen X-Chromosom die Stilllegung dieses Gens fördert, verfügen XX-Mäuse insgesamt über weniger Tlr7 als XY–-Mäuse, deren Nervensystem folglich stärker degeneriert.
Ich mach mir die Maus, widewide wie sie mir gefällt?
Es gibt noch radikalere Versuche, den störenden Einfluss der Sexualhormone auf die Analyse der geschlechtschromosomalen Effekte auszuschließen: In den Embryonen der sogenannten SF1 knockout mouse verhindert eine Mutation im Gen SF1 (für steroidogenic factor 1) die Bildung von Keimdrüsen- und Nebennieren-Anlagen. Auch Teile des Hypothalamus und Hypophyse entwicklen sich nicht normal. Vor allem wegen der fehlenden Nebennierenrinde, dem Produktionsort der Corticosteroide, sterben die Tiere kurz nach der Geburt – es sei denn, man injiziert ihnen sofort Glucocorticoide und implantiert ihnen dann Nebennierenrinden-Gewebe aus intakten Mäusen.
Auch bei diesen Kreaturen unterscheiden sich die „Geschlechter“ (sofern man keimdrüsen- und sexualhormonlose Tiere mit XX- und mit XY-Chromosomen so nennen möchte) noch ein wenig, unter anderem im Körpergewicht und in der Expression eines Enzyms in einigen limbischen Gehirnstrukturen. Ob auch ihr Immunreaktionen Unterschiede aufweist, weiß ich nicht. Aber die Ergebnisse wären ohnehin sehr schwer zu interpretieren, denn diese Geschöpfe haben so viele Defekte, dass sich aus ihnen m. E. nicht viel über normale Säugetiere lernen lässt.
Und als wäre das alles nicht schon kompliziert und entmutigend genug, gibt es ein weiteres Mausmodell für Multiple Skerose, den C57BL/6-Stamm, bei dem die experimentelle Autoimmun-Enzephalitis bei Männchen und Weibchen und auch bei kastrierten Tieren mit allen vier Kern-Genotypen gleich häufig auftritt.
Zusammenfassung: Es ist kompliziert
Man kann also allerlei Mäuse züchten, Puzzle-artig zusammensetzen, zurichten, retten und opfern, um die normalerweise eng verwobenen, zum Teil gegenläufigen Effekte von Sexualhormonen, Geschlechtschromosomen-Genen und epigenetischen Markierungen dieser Gene voneinander zu trennen. Solche Experimente können beliebig kompliziert und artifiziell werden, liefern aber wichtige Hinweise auf mögliche Gründe für Geschlechtsunterschiede bei Autoimmunerkrankungsrisiken.
Leider lautet die Hauptbotschaft dieser Versuche: Es gibt keine einfache Antwort. Sexualhormone wirken sowohl lang- als auch kurzfristig, und zwar unter anderem auf das Immunsystem und auf das Gehirn. Die Geschlechtschromosomen beeinflussen ebenfalls verschiedene Systeme, darunter wiederum Immunsystem und Gehirn, und wirken den Sexualhormonen zum Teil entgegen. Darüber hinaus wirken sie auch von Organ zu Organ unterschiedlich – und über verschiedene Mechanismen, etwa X-Dosis und parentales Imprinting. Bei einigen Mausmodellen für Autoimmunerkrankungen scheinen diese Mechanismen jedoch keine Rolle zu spielen – oder sich gegenseitig aufzuheben. Und das heißt: Man muss eben doch jede Autoimmunerkrankung einzeln untersuchen, an Mäusen, aber auch an Menschen.
Literatur:
Ältere Blogbeiträge mit Literaturzusammenfassungen: