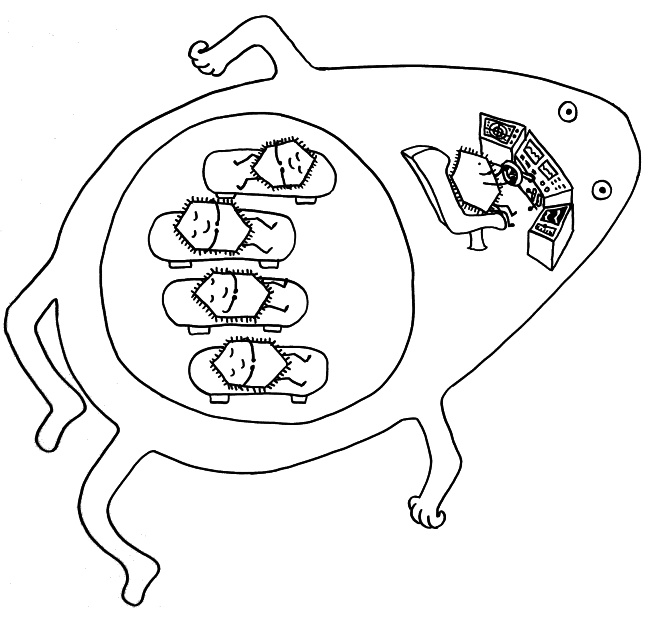
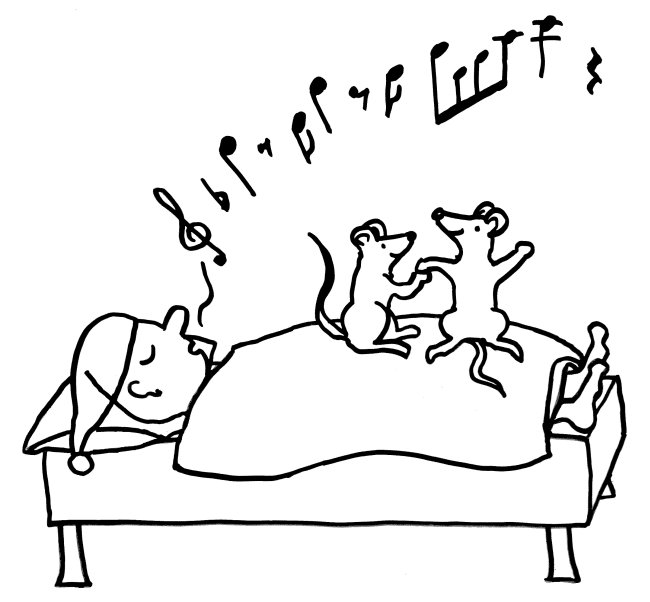
Im letzten Beitrag habe ich eine Studie vorgestellt, der zufolge unreife rote Blutkörperchen unser Immunsystem in den Wochen nach der Geburt so stark zäumen, dass die Erstbesiedlung des Darms mit gutartigen Bakterien nicht zu einer gefährlichen großflächigen Entzündung führt. Hier nun die passenden Skizzen – zunächst ein erwachsener, kernloser Erythrozyt, der bekanntlich die Aufgabe hat, Sauerstoff aus den Lungen in unser Gewebe zu transportieren, und ein junger, unreifer Erythrozyt, der wegen seines Zellkerns noch nicht die typische Scheibenform der roten Blutkörperchen angenommen hat. Seine Aufgabe ist es, Immunreaktionen aufzuhalten:
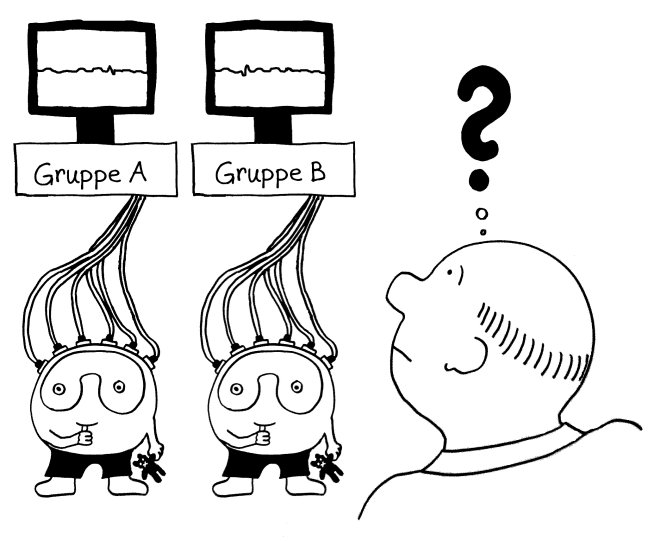
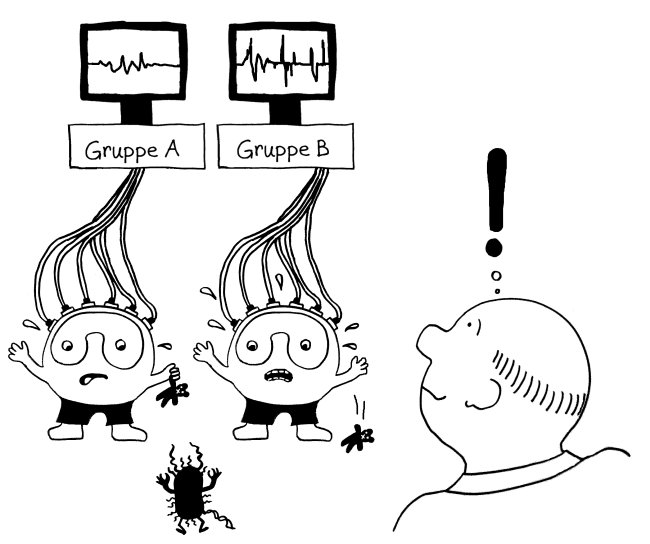
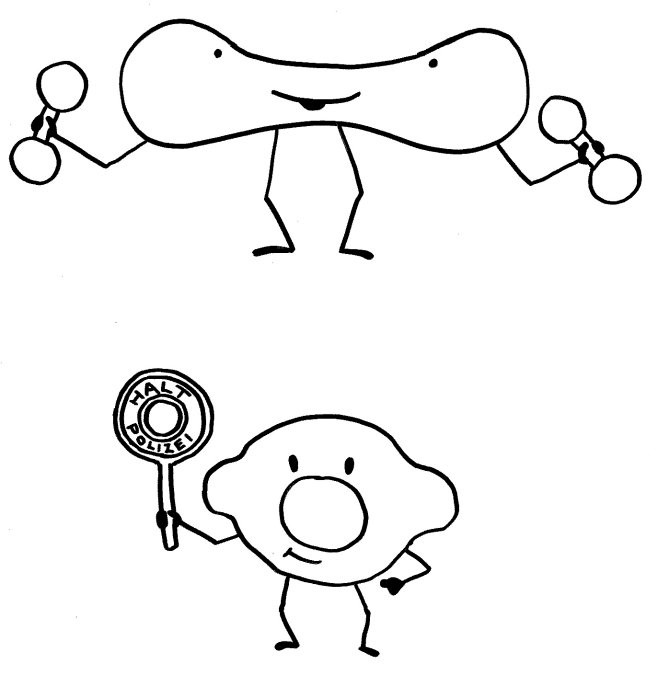 Dass die kernhaltigen rote Blutkörperchen von Nicht-Säugetieren wie Fischen und Vögeln auch Aufgaben im Immunsystem übernehmen, ist schon lange bekannt. Insofern sollte es uns nicht überraschen, dass dies auch bei Menschen der Fall ist – wenn auch nur in einem schmalen Zeitfenster: Vorläufer späterer roter Blutkörperchen, die den Marker CD71 auf der Oberfläche tragen, hemmen durch Enzyme und womöglich weitere lösliche Substanzen die Aktivität der T-Zellen, B-Zellen, dendritischen Zellen und Makrophagen von Neugeborenen. Eventuell fördern sie zudem durch Freisetzung von Zytokinen die Bildung von regulatorischen T-Zellen (Tregs) und T-Helferzellen des Typs 2 (Th2).
Dass die kernhaltigen rote Blutkörperchen von Nicht-Säugetieren wie Fischen und Vögeln auch Aufgaben im Immunsystem übernehmen, ist schon lange bekannt. Insofern sollte es uns nicht überraschen, dass dies auch bei Menschen der Fall ist – wenn auch nur in einem schmalen Zeitfenster: Vorläufer späterer roter Blutkörperchen, die den Marker CD71 auf der Oberfläche tragen, hemmen durch Enzyme und womöglich weitere lösliche Substanzen die Aktivität der T-Zellen, B-Zellen, dendritischen Zellen und Makrophagen von Neugeborenen. Eventuell fördern sie zudem durch Freisetzung von Zytokinen die Bildung von regulatorischen T-Zellen (Tregs) und T-Helferzellen des Typs 2 (Th2).
Shokrollah Elahi vermutet, dass die massiven Entzündungen, unter denen viele Frühgeborene leiden, auf einen Mangel an CD71+-Zellen zurückzuführen sind. Diese Schutzpolizisten entstehen nämlich vor allem in den letzten Schwangerschaftswochen vor dem normalen Geburtstermin. Bei einer Frühgeburt ist ihre Zahl noch viel zu gering, um das Immunsystem während der Erstbesiedlung des Darms mit unseren Darmbakterien vom Amoklauf abzuhalten.
Wie aber werden unreife Erythrozyten „erwachsen“? Sie versammeln sich im roten Knochenmark um Makrophagen, scheiden ihre Zellkerne ab und nehmen ihre Arbeit als Sauerstofftransporteure auf. Die Kerne, die dabei nur stören würden, werden von den Makrophagen vertilgt:
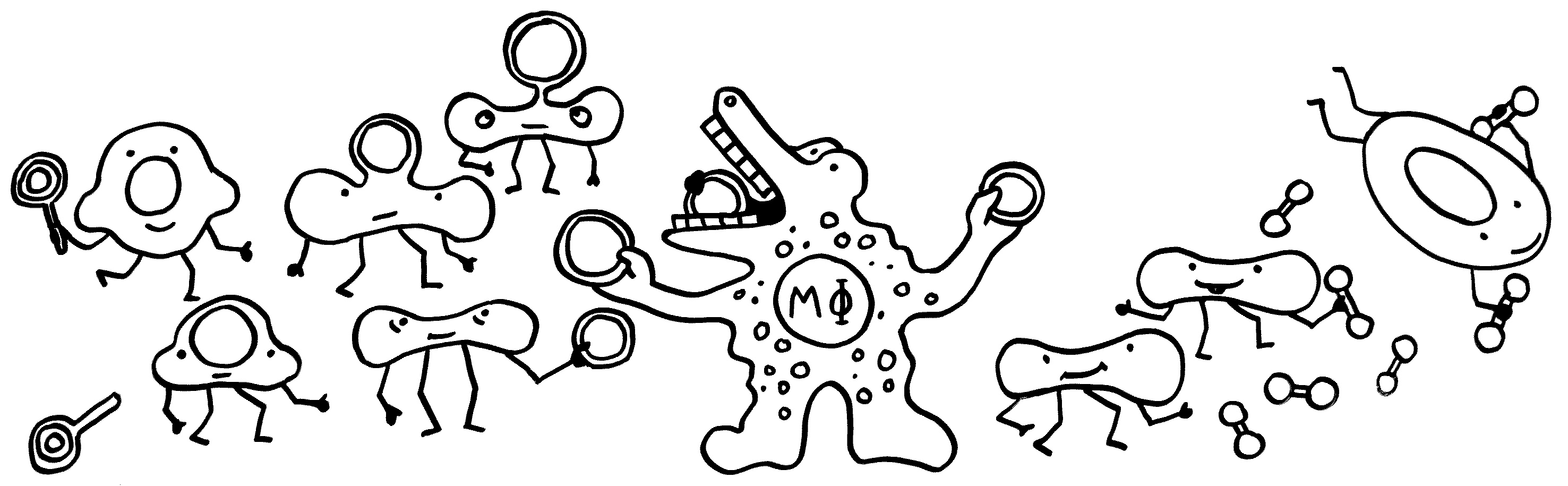
Wie so oft übernehmen die Makrophagen also die Müllentsorgung – besonders wichtig, wenn es um die Beseitigung von Kernen geht, da diese jede Menge Nukleinsäuren (DNA) enthalten, die andernfalls starke Immunreaktionen auslösen würden. Extrazelluläre Nukleinsäuren deuten nämlich normalerweise auf Infektionen oder ein massives Zellsterben hin.
Lit.: S. Elahi (2014): New insight into an old concept: role of immature erythroid cells in immune pathogenesis of neonatal infection