Glucocorticoide sind Steroidhormone, die – neben vielen weiteren Wirkungen – Immunreaktionen und damit auch Entzündungen hemmen. In unserem Körper übernimmt vor allem das sogenannte Stresshormon Cortisol (umgangssprachlich: Kortison) diese Aufgabe, aber man hat etliche weitere dieser Hormone erzeugt, die stabiler sind und stärker wirken, unter anderem das neulich erwähnte Dexamethason oder auch Prednison. Ihren Namen verdanken die Glucocorticoide ihrem Einfluss auf den Glucose-Stoffwechsel, ihrem Entstehungsort, der Nebennierenrinde (lateinisch Cortex glandulae suprarenalis), und ihrer Zugehörigkeit zur Stoffklasse der Steroide.
Obwohl ihre entzündungshemmende Wirkung schon lange bekannt ist und therapeutisch bei zahlreichen Erkrankungen (etwa Autoimmunerkrankungen oder chronischen Entzündungen) und nach Operationen genutzt wird, hat die Wissenschaft lange nicht so recht verstanden, auf welchen Weg diese Hormone Entzündungen hemmen. Nachdem man Glucorticoid-Rezeptoren im Zytoplasma entdeckt und in den 1980er-Jahren kloniert hatte, hat man sie sehr gründlich in allerlei Experimenten untersucht und sich folgende Erklärung zurechtgelegt: Das Hormon dringt durch die Zellmembran ins Zytoplasma ein und trifft dort auf seinen Rezeptor, der mit verschiedenen weiteren Proteinen dort verankert und inaktiv gehalten wird. Sobald das Hormon nach dem Schlüssel-Schloss-Prinzip an seinen Rezeptor bindet, werden die Begleitproteine durch Konformationsänderungen abgetrennt, und der Rezeptor wacht gewissermaßen auf.
Als Monomer, also als einzelner aktivierter Rezeptor, interagiert er im Zytoplasma und in den Organellen mit allen möglichen Strukturen. Vor allem aber wandert er als sogenannter Liganden-aktivierter nukleärer Transkriptionsfaktor zusammen mit dem Glucocorticoid durch die Kernporen in den Zellkern ein und lagert sich dort paarweise (als sogenanntes Dimer) an spezifische DNA-Sequenzen an. Dadurch regt er entweder die Ablesung von Genen an, die entzündungshemmende Proteine wie Interleukin-10 codieren, oder er blockiert die Ablesung anderer Gene, deren Produkte proinflammatorisch wirken. Darüber hinaus kann er die Wechselwirkung der Histone (also der „DNA-Kabelspulen“) und anderer Proteine mit der DNA im Zellkern beeinflussen und so auch indirekt die Ablesung von Genen verändern. Die Lehrbücher und das Internet sind voll von wissenschaftlichen und populärwissenschaftlichen Schemazeichnungen, in denen diese Abläufe mehr oder weniger vereinfacht dargestellt sind, etwa in einer Übersichtsarbeit von 2024 oder als Adobe Stock Image.
Der Itaconat-Weg
Immer wieder sprachen aber Versuchsergebnisse dagegen, dass dies der einzige Mechanismus ist, über den Glucocorticoide im Zellkern die Ablesung entzündungsfördernder Gene hemmen und die Ablesung entzündungshemmener Gene fördern. 2024 gab es einen Durchbruch: Ein Forschungsteam um Jean-Philippe Auger und Gerhard Krönke (Nature-Publikation hinter Bezahlschranke) klärte einen weiteren Wirkungsweg auf, der auch unter Stoffwechsel- und Evolutionsaspekten ziemlich einleuchtend ist – und daher auch in Band 2 meines Buches eingeht. Er führt über die Mitochondrien aktivierter Makrophagen.
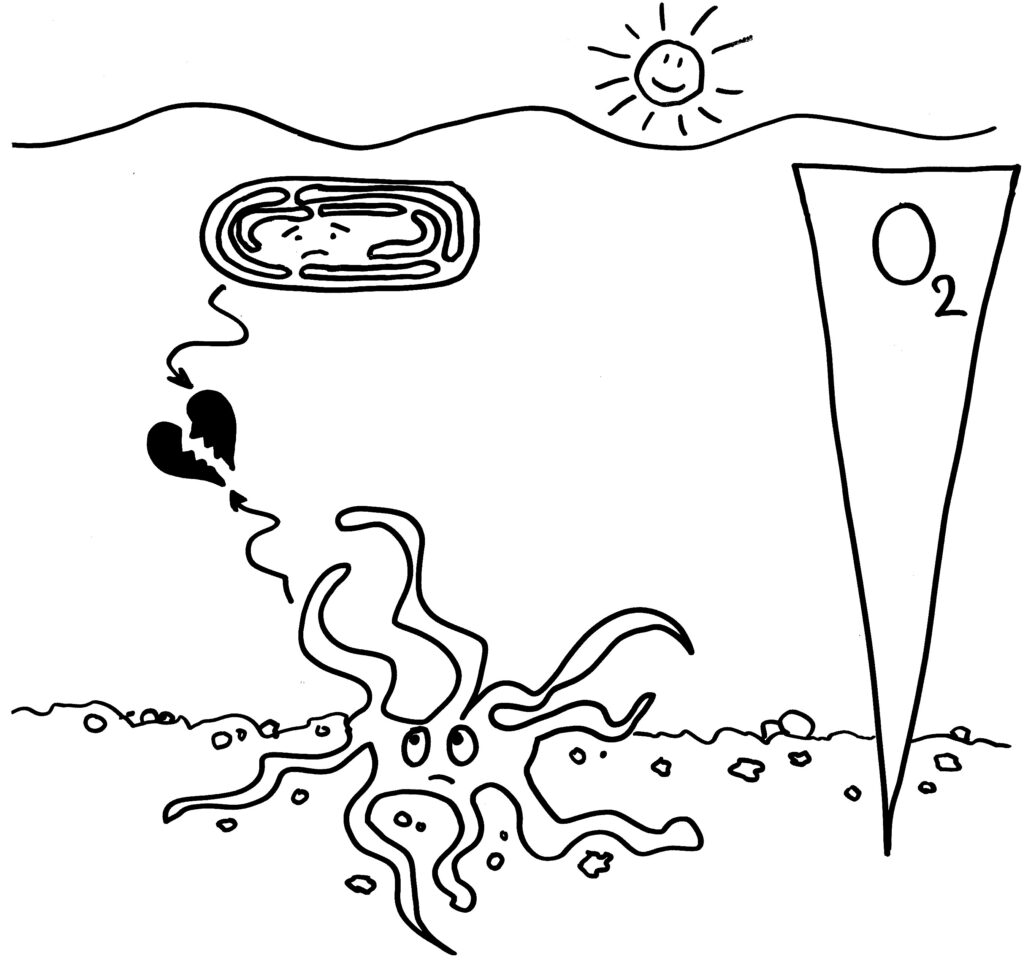
In diesen Organellen, die – siehe Titelbild! – gerne als Kraftwerke der Zellen bezeichnet werden, wird aus unserer Nahrung Energie generiert – vor allem in Form von ATP-Molekülen, die dann in der ganzen Zelle biochemische Reaktionen oder Transportprozesse befeuern, die eine Energiezufuhr benötigen. Diese Energiegewinnung verläuft über drei jeweils mehrschrittige Teilprozesse:
- Noch außerhalb der Mitochondrien, im Zytosol, findert die Glykolyse statt. Dabei wird Glucose (6 Kohlenstoffatome) zu 2 Pyruvat-Molekülen (3 Kohlenstoffatome) zerlegt, die von einem Enzymkomplex ins Innere der Mitochondrien transportiert werden.
- In den Mitochondrien wird Pyruvat zu Acetyl-CoA (2 Kohlenstoffatome) und Kohlendioxid zerlegt. Auch andere Stoffwechselvorgänge wie der Abbau von Fettsäuren können Acetyl-CoA liefern. Im Citrat- oder Tricarbonsäure-Zyklus wird dieses dann zu Kohlendioxid und Wasser abgebaut; dabei werden energiereiche Elektronen in den Molekülen NADH und FADH2 gebunden. (Dieser Zyklus kann unter bestimmten Umweltbedingungen auch rückwärts verlaufen.)
- In der Atmungskette, die in der inneren Mitochondrien-Membran abläuft, wird durch schrittweise Übertragung der energiereichen Elektronen aus NADH und FADH2 ein Protonengradient über die Membran aufgebaut, der schließlich zur Gewinnung von ATP genutzt wird – ein Vorgang, den man sich wie die Stromgenerierung an einem Kraftwerk in einer Staumauer vorstellen kann, bei der ja ebenfalls ein Gradient (Höhenunterschied des Wassers zu beiden Seiten der Mauer) zur Energiegewinnung ausgenutzt wird.
Für uns ist hier der zweite, zyklische Stoffwechselweg entscheidend. Eines seiner frühen Zwischenprodukte ist die Aconitsäure oder cis-Aconitat. Normalerweise wird dieses gleich weiter zu cis-Citrat umgewandelt. Anders in sogenannten aktivierten Makrophagen: Behandelt man diese Immunzellen mit Lipopolysacchariden oder LPS (einem hochwirksamen Antigen aus der Hülle gramnegativer Bakterien, das die antibakterielle Abwehr stimuliert) plus Gamma-Interferon (einem an Entzündungsdreaktionen, etwa nach einer Virusinfektion, beteiligten Zytokin), so wechseln die Makrophagen in den Kampfmodus. Unter anderem stellen sie das Enzym Aconitat-Decarboxylase 1 (ACOD1) her, das in ihre Mitochondrien einwandert. Dort wandelt es Aconitat zu einem Nebenprodukt des Citrat-Zyklus um: Itaconat. (Ja, der Entdecker dieser Reaktion hat das Reaktionsprodukt einfach durch Umstellen der Buchstaben von Aconitat benannt!)
Itaconat wirkt auf mehreren Wegen entzündungshemmend – was zunächst pradox wirkt, da aktivierte Makrophagen ja eigentlich Infektionen und andere Störungen bekämpfen sollen, wozu sie eine Entzündungsreaktion unterstützen sollten. Das tun sie auch; ich gehe im nächsten Artikel darauf ein. Aber damit die Entzündung rechtzeitig beendet wird, entsteht Itaconat. Dieses Molekül ist ein klassischer Imunmetabolit: ein Stoffwechselprodukt, das zugleich als Signal innerhalb des Immunsystems wirkt. Es ist immunmodulierend, und zwar auf vielfältige Weise. So blockiert es ein Enzym in den Makrophagen-Mitochondrien, dessen Aktivität die Produktion entzündungsfördernder Zytokine und reaktiver Sauerstoffspecies fördert, die als Stress-Signale ebenfalls Entzündungen anheizen. Und es blockiert ein anderes Enzym, das normalerweise im Zellkern die Ablesung von Genen beeinflusst; auch das bremst Entzündungen.
Itaconat kann die Makrophagen auch verlassen und sich im Serum ansammeln. Von dort aus können weitere immunzellen es aufnehmen. So hat man festgestellt, dass Itaconat (bzw. künstlich hergestellte Itaconat-Derivate, die leichter zu erforschen sind) die T-Zell-Differenzierung verschiebt: Es entstehen mehr entzündungshemmende regulatorische T-Zellen (Tregs) und weniger entzündungsfördernde Th17-Zellen.
Alternative zur Glucocorticoid-Therapie?
Und jetzt kommt’s: Neben LPS, Gamma-Interferon und anderen Entzündungstriggern wie Sport können auch Glucocorticoide die Itaconat-Produktion in den Mitochondrien von Makrophagen stark erhöhen – und zwar in bereits aktivierten Makrophagen. Die Steroidhormone binden an einen Rezeptor im Zytosol, der dort das Enzym Pyruvatdehydrogenase (PDH) festhält. Durch die Bindung des Hormons löst sich der Rezeptor vom Enzym, und dieses kann in die Mitochondrien einwandern, wo es den Citratzyklus beeinflusst und damit die Itaconat-Produktion erhöht.
Glucocorticoide rufen gerade bei längerer, systemischer Gabe viele Nebenwirkungen hervor, die zum Teil so schwer werden, dass die Therapie angebrochen werden muss. Daher hat der neu entdeckte Clucocorticoid-Wirkungsweg über das Itaconat aus den Mitochondrien aktivierter Makrophagen großes Interesse geweckt: Vielleicht lassen sich stabile Itaconat-Derivate entwickeln, die die entzündungshemmende Wirkung beibehalten, aber zugleich weniger schwere Nebenwirkungen mit sich bringen.
Aber Vorsicht: Itaconat ist gewiss kein Wundermittel. Während es bei chronischen Entzündungen und Autoimmunerkrankungen womöglich die überschießenden Immunreaktionen einhegen kann, scheint es bei Krebs und Sepsis eher zu schaden. Es behindert zum Beispiel Immunreaktionen, die Tumorzellen zum Absterben bringen sollen. Und bei einer Sepsis scheint es die aus dem Ruder gelaufene Entzündung sogar zu verstärken, statt sie zu beenden. Bis zu einem möglichen Einsatz wird daher noch viel Pyruvat den Citratzyklus hinunterfließen.
Literatur:
Studie in Nature entschlüsselt, wie Kortison Entzündungen dämpft (PM zu Auger et al., 2024)
Yin Luo et al. (2024): Metabolic Regulation of Inflammation: Exploring the Potential Benefits of Itaconate in Autoimmune Disorders
Eva M. Pålsson-McDermott, Luke A.J. O’Neill (2025): Gang of 3: How the Krebs cycle-linked metabolites itaconate, succinate, and fumarate regulate macrophages and inflammation
Maria Tada, Michihito Kono (2025): Metabolites as regulators of autoimmune diseases
Yifan Xie (2025): Itaconate: A Potential Therapeutic Strategy for Autoimmune Disease

 Die Frage ist uralt: Wie finden Tauben, die man weit von ihrem Schlag entfernt aussetzt, nach Hause? Mein letzter Stand war, dass es etwas mit magnetischen Partikeln in den Schnäbeln der Vögel zu tun haben könnte, aber diese Hypothese ließ sich offenbar nicht eindeutig bestätigen.
Die Frage ist uralt: Wie finden Tauben, die man weit von ihrem Schlag entfernt aussetzt, nach Hause? Mein letzter Stand war, dass es etwas mit magnetischen Partikeln in den Schnäbeln der Vögel zu tun haben könnte, aber diese Hypothese ließ sich offenbar nicht eindeutig bestätigen.