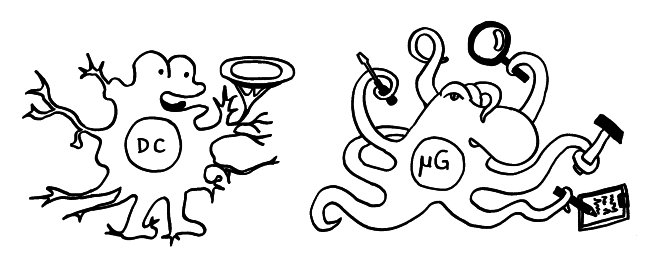
Telomere und ihre Rolle bei der Alterung (Seneszenz) von Immunzellen habe ich im Blog schon öfter thematisiert, denn ihre Verkürzung bei jeder Zellteilung könnte einer der Faktoren sein, die bei älteren Menschen zum Ausbruch von Autoimmunerkrankungen beitragen. Jetzt gibt es sensationelle Neuigkeiten:
Lanna, A., Vaz, B., D’Ambra, C. et al. An intercellular transfer of telomeres rescues T cells from senescence and promotes long-term immunological memory. Nat Cell Biol (2022). https://doi.org/10.1038/s41556-022-00991-z
Bei Nature in der Artikel nur in einer Leseansicht ohne Markierungs- und Download-Funktion verfügbar, aber das Manuskript ist bei biorxiv zu finden. Frei zugänglich ist auch die Meldung bei The Scientist: T Cells Ward Off Aging with Help from Their Friends
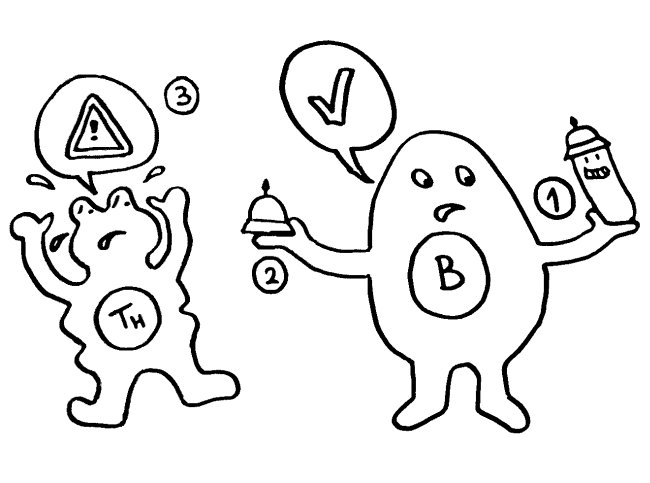
Abstract: T-Zellen sollen nach bisheriger Vorstellung ihre Alterung aufhalten durch Telomerase, die ihre Telomere wieder verlängert (s. Nothing in Oncology Makes Sense Except in the Light of Evolution). Die Autor*innen zeigen hier: Vor allem naive T-Zellen und zentrale Gedächtnis-T-Zellen nehmen Telomer-Vesikel von APCs auf und verlängern ihre Telomere so ohne Telomerase-Aktivität. Bei Kontakt bauen APCs Shelterin ab; ihre Telomere werden vom Trimming-Faktor TZAP abgeschnitten und an der Immun-Synapse in extrazelluläre Vesikel verpackt. Diese enthalten auch den Rekombinationsfaktor Rad51, der die Fusion mit Enden der T-Zell-Telomere bewirkt. Die antigenspezifischen T-Zellen empfangen die Telomerverlängerungen (im Mittel ca. 3000 Basenpaare) vor ihrer klonalen Expansion, was zu langfristiger Immunität führt.
Intro: Telomere = TTAGGG-Wiederholungen. Bei kurzen Telomere von < 4kb lässt die Teilungsfähigkeit nach (replikative Seneszenz). Bei vielen Alterskrankheiten und Krebs werden kurze Telomere beobachtet. Zellen können die Verkürzung mit oder ohne Telomerase aufhalten. – Immunologische Synapsen dienen der Kommunikation zwischen APCs und Lymphozyten, initiieren Immunreaktionen, die zu langlebigen Gedächtnis-T-Zellen führen. Eine Synapse aktiviert die Telomerase in der T-Zelle zunächst; mehrfache synaptische Interaktion zieht aber einen Aktivitätsrückgang nach sich und damit die Seneszenz der T-Zelle, ein Nachlassen des immunologischen Gedächtnisses, u. U. also mehr Infektionen, Krebs, Tod. Telomerase allein kann also die T-Zell-Seneszenz letztlich nicht verhindern. Wenn T-Zellen aber Telomere aus APCs empfangen, werden sie zu Stammzell-ähnlichen und/oder zentralen langlebigen Gedächtniszellen, während andere T-Zellen altern und sterben. Die APCs entscheiden also bei der ersten Synapse über das Schicksal der T-Zellen.
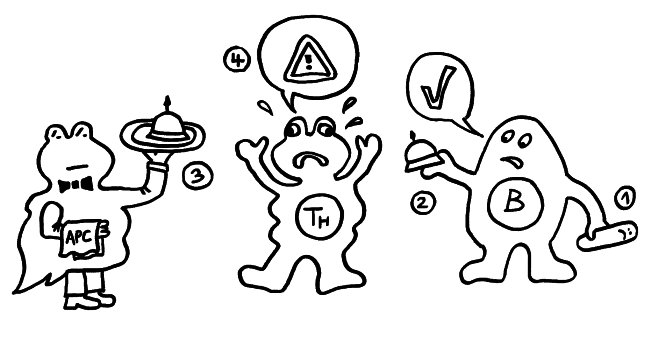
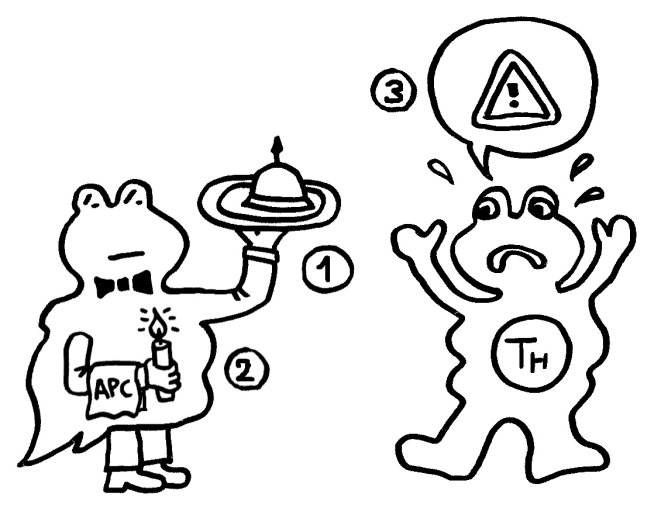
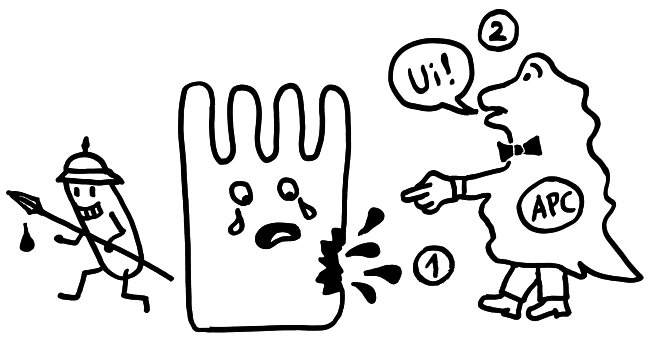
Ergebnisse: Das Team hat eine Verlängerung von T-Zell-Telomeren und gleichzeitige Verkürzung von APC-Telomeren beobachtet, in Gegenwart von Antigenen aus Epstein-Barr-Virus-, Influenza-Virus- und Cytomegalovirus-Lysaten. Das klappt auch in T-Zellen ohne Telomerase (Knock-out), kann auch nicht auf alternativem Telomer-Verlängerungs-Mechanismus per Rekombination und DNA-Synthese beruhen, weil sich die T-Zellen noch gar nicht teilten. Gelabelte Telomer-DNA aus den APCs tauchte später in den T-Zellen auf; damit war die Herkunft belegt. TCR-besetzte planare Lipiddoppelschichten lösen die Telomer-Freisetzung aus den APCs ebenso gut aus wie komplette T-Zellen. Außer TCR sind auch Anti-CD3 und Antigene auf den APC-MHC-Komplexen nötig. Die APCs sterben nicht nach Telomer-Angabe, zeigen auch kein Blebbing. Myeloide APCs (dendritische Zellen und Monozyten) geben am meisten Telomere ab, B-Zellen weniger. Die Vesikel enthalten außer Telomeren auch Histokompatibilitäts-Antigen-Proteine sowie TZAP (telomeric zinc-finger associated protein), ein telomerbindendes Protein, das die terminalen Enden von Chromosomen beschneidet (Telomer-Trimming) und fürs Abschneiden und Verpacken der Telomere nötig ist. Damit TZAP an die Telomere binden kann, muss das Shelterin herunterreguliert und abgebaut werden, das die Telomere normalerweise stabilisiert und vor DNA-Reparaturmechanismen beschützt. Die Vesikel enthalten auch Rad51, einen homologen Rekombinationsfaktor, der an der Telomer-Verlängerung beteiligt ist und für den Anbau der gestifteten Telomere in den T-Zellen nötig ist. In Mäusen wandern die T-Zellen mit den APC-verlängerten Telomeren rasch in Langzeit-Überlebensnischen wie Milz und Lymphknoten; im Blut sind sie kaum zu finden.
Influenza-Impf-Experiment: Mäusen wurden antigenspezifische, geprimete T-Zellen mit APC-verlängerten oder mit nicht verlängerten Telomeren injiziert; die Kontrollgruppe erhielt keine solchen T-Zellen. Entweder 18 Stunden oder 15 Tage nach der Injektion wurden die Tiere mit Influenza infiziert. Ungeimpfte Mäuse starben alle rasch; mit unveränderten Telomer-T-Zellen wurde die frühe Infektion gut abgewehrt (alle Tiere überlebten), die späte Infektion aber nicht (alle Tiere starben im Lauf einige Tage), weil kein Gedächtnis ausgebildet wurde. Mäuse mit T-Zellen mit APC-verlängerten Telomeren überlebten sowohl eine frühe als auch eine späte Infektion; das immunologische Gedächtnis reichte aus.
Diskussion: Es gibt auch andere Pfade zu Gedächtnis-T-Zellen, teils von naiven T-Zellen ausgehend, teils nach Effektor-Tätigkeit durch Umschalten von Glykolyse auf oxidative Phosphorylierung. Vermutung: Die Antigenstärke könnte die Menge der übertragenen Telomere und damit die Zahl der möglichen nachfolgenden Zellteilungen beeinflussen. Aber auch bei identischen Antigenen hat ein Großteil der T-Zellen keine Telomere aufgenommen; sie blieben kurzlebige Effektorzellen. T-Zellen sollen nach wiederholter Antigenstimulation seneszent werden; stark ausdifferenzierte Effektor-T-Zellen können ihre Telomerase nicht weiter aktivieren; ihre Proliferation lässt nach -> lineare Seneszenz. Alternatives Modell: Die Unfähigkeit, während der Antigenstimulation APC-Telomere zu empfangen, besiegelt schon das Schicksal der T-Zellen -> Seneszenz. Nach Auflösung der Synapse startet die massive Proliferation; jetzt kommt die Telomerase hinzu, die an alle Chromosomen pro Teilung ca. 100-200 bp anhängt. Ein Telomer-Transfer verlängert dagegen bestimmte, vermutlich sehr kurze, Telomere um ca. 3000 bp noch vor den Zellteilungen. Vermutlich ist der Telomer-Transfer das schon länger postulierte Signal, von dem die terminale Differenzierung der T-Zellen abhängt. Unklar bleibt, wonach sich entscheidet, ob eine T-Zelle die Telomere einbauen kann. – Darüber hinaus kann es weitere Wege zur Bildung von Gedächtniszellen geben, etwa eine Dedifferenzierung von Effektorzellen, sodass sie ebenso Stammzell-ähnlich werden wie die Gedächtniszellen mit den APC-Telomeren.
Abb. 8: Versuchsdesign mit Absterben der grippeinfizierten Mäuse ohne injizierte T-Zellen mit verlängerten Telomeren während der Gedächtnis-Phase; in der Effektorphase haben die Tiere eine Infektion noch überlebt. Mäuse mit injizierten T-Zellen mit verlängerten Telomeren überleben auch eine späte Infektion während der Gedächtnis-Phase, weil die T-Zellen so lange leben. Außerdem Illustration der Synapse mit Vesikeltransfer und Telomerfusion.