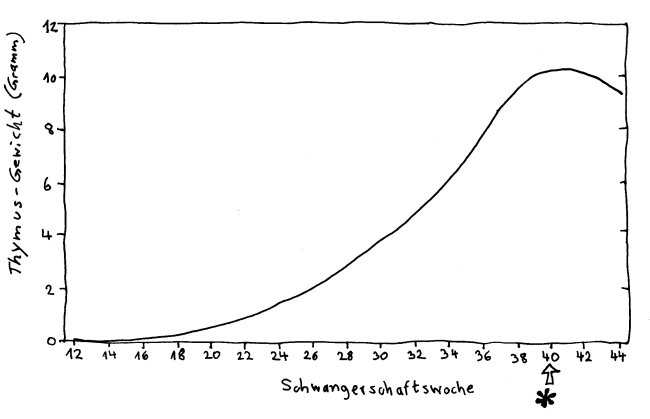
Der Thymus ist ein unauffälliges Organ zwischen Brustbein und Herz; die meisten Menschen wissen nichts von seiner Existenz. Dabei kommt nicht um ihn herum, wer das Grundprinzip der sogenannten erworbenen Immunität verstehen will:
So, wie die Evolution nicht zielgerichtet voranschreitet, sondern über das Wechselspiel von Zufallsvarianten und Auslese, so richtet sich auch die erworbene Abwehr nicht gezielt gegen Krankheitserreger und andere Gefahren, sondern stellt einfach eine gigantische Auswahl an Rezeptor-Zufallsvarianten zur Verfügung, um auf alles zu reagieren, was nicht in den Körper hineingehört. Damit der Körper sich dabei nicht selbst bekämpft (was bei Autoimmunerkrankungen passiert), müssen alle T-Zellen, deren Rezeptoren stark an körpereigene Antigene binden, rechtzeitig ausgeschaltet oder zu Friedensstiftern umgebaut werden.
Anders geht es logischerweise nicht: Unser Körper weiß a priori nichts über die Welt und ihre Gefahren, die sich ja ständig verändern. Er weiß nur etwas über sich selbst; also bildet er die Differenz aus allen erdenklichen Erkennungsmustern und den Erkennungsmustern für die eigenen Bestandteile. Beide Aufgaben – die Generierung der immensen Vielfalt an T-Zell-Rezeptoren und die Eliminierung der stark autoreaktiven T-Zellen, die zu Autoimmunreaktionen führen würden – übernimmt der Thymus.
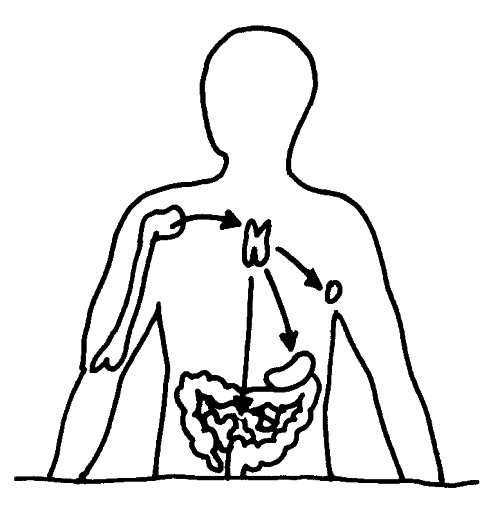
Aus dem Knochenmark wandern ständig unreife T-Zellen oder Thymozyten in den Thymus ein, wo sie durch die sogenannte somatische Rekombination oder V(D)J-Rekombination aus einem begrenzten Satz an Genen eine Vielzahl individueller Rezeptoren erzeugen, die dann auf ihre Antigen-Bindungsfähigkeit getestet werden. Die dabei nicht aus dem Verkehr gezogenen, nunmehr reifen T-Zellen wandern aus dem Thymus in die anderen Teile des Lymphsystems, etwa die Milz, die Lymphknoten oder das Lymphgewebe des Verdauungstrakts, wo sie ihre Umgebung überwachen und Alarm schlagen, sobald ihre Rezeptoren an die passenden Antigene binden.
Die Entwicklung der künftigen T-Zellen im Thymus durchläuft mehrere Phasen, die teils mit starker Vermehrung durch Zellteilung, teils mit einem Rückgang der Zellzahl durch Verkümmern beim Ausbleiben von Überlebenssignalen oder durch die Einleitung eines kontrollierten Zelltods einhergehen. Anfangs sind die Thymozyten „doppelt negativ“ (DN), denn ihre Oberfläche weist noch keinen der beiden Korezeptoren CD4 – typisch für T-Helferzellen – und CD8 – typisch für zytotoxische T-Zellen – auf. (Welche Aufgabe diese Korezeptoren haben und wie ein T-Zell-Rezeptor aufgebaut ist, habe ich hier in der dritten Zeichnung dargestellt.)
Am Ende der ersten Vermehrungsphase werden zunächst die Gene für eine Hälfte des T-Zell-Rezeptors, β-Kette genannt, durch das Herausschneiden von Zwischensequenzen neu zusammengesetzt oder rekombiniert. Die daraufhin produzierte Rezeptorhälfte wird an einen provisorischen Platzhalter (pα) gekoppelt, der später durch die andere Rezeptorhälfte – die α-Kette – ersetzt wird. Nur diejenigen Thymozyten, deren β-Rezeptorhälften funktionstüchtig sind, also korrekt in die Zelloberfläche eingebaut werden und von dort Signale ins Zellinnere leiten können, erhalten von ihrer Umgebung im Thymus ein Überlebenssignal; die übrigen verkümmern im Rahmen der sogenannten β-Selektion.
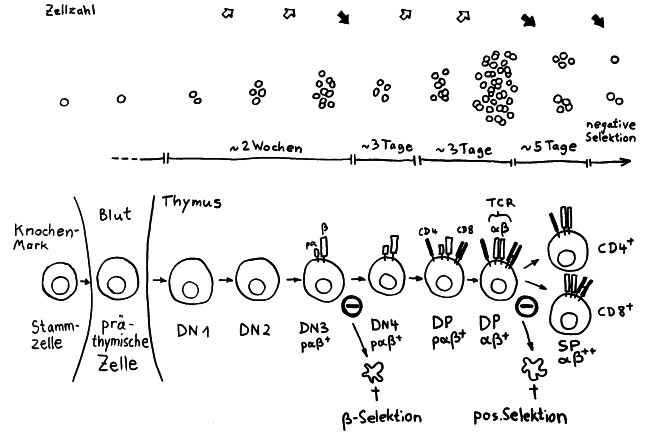
Anschließend vermehren sich die überlebenden T-Zell-Vorläufer stark, und sie fangen an, beide Korezeptoren CD4 und CD8 herzustellen. Daher heißen sie nun „doppelt positiv“ (DP). Jetzt werden auch die Gene für die α-Kette des T-Zell-Rezeptors durch somatische Rekombination neu zusammengeschnitten, um ein möglichst breites Spektrum an Antigen-Bindungsfähigkeiten zu erzeugen.
Die DP-Thymozyten haben drei Tage Zeit, um einen funktionsfähigen T-Zell-Rezeptor herzustellen. Gelingt ihnen das nicht, so gehen sie durch das Ausbleiben von Überlebenssignalen aus ihrer Umgebung ein. Haben sie einen intakten Rezeptor erzeugt, der mehr oder weniger gut an die Antigen-MHC-Komplexe bindet, die ihm von den kortikalen Thymus-Epithelzellen (cTEC) präsentiert werden, so überleben sie diese sogenannte positive Selektion und wandern aus der Thymus-Rinde ins Thymus-Mark weiter. Dort müssen die Zellen, die mittlerweile nur noch einen der beiden Korezeptoren herstellen und damit einfach positiv (single positive = SP) sind, noch die negative Selektion bestehen, in der autoreaktive T-Zellen ausgesondert werden. Dazu in einem der folgenden Beiträge mehr; wir verweilen heute in Kortex, in der Rinde des Thymus.
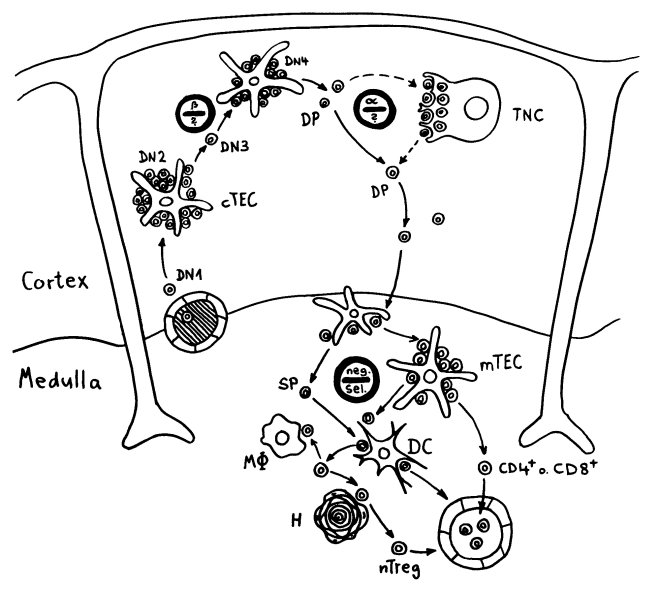
Bereits in den 1980er-Jahren fiel den Forschern eine Kuriosität auf: Ein Teil der kortikalen Thymus-Epithelzellen stellt offenbar das Hormon Oxytocin her; jedenfalls wird das Oxytocin-Gen in ihnen abgelesen. Und die doppelt positiven Thymozyten stellen einen Oxytocin-Rezeptor her und reagieren auf Oxytocin mit starker Zellteilungsaktivität. Alles deutete also auf eine hormonelle Kommunikation zwischen diesen beiden Zelltypen hin. Seltsamerweise ließ sich aber in der Flüssigkeit aus den Hohlräumen zwischen den Thymuszellen selbst mit sehr empfindlichen Nachweismethoden nie auch nur der leiseste Hauch Oxytoxin aufspüren.
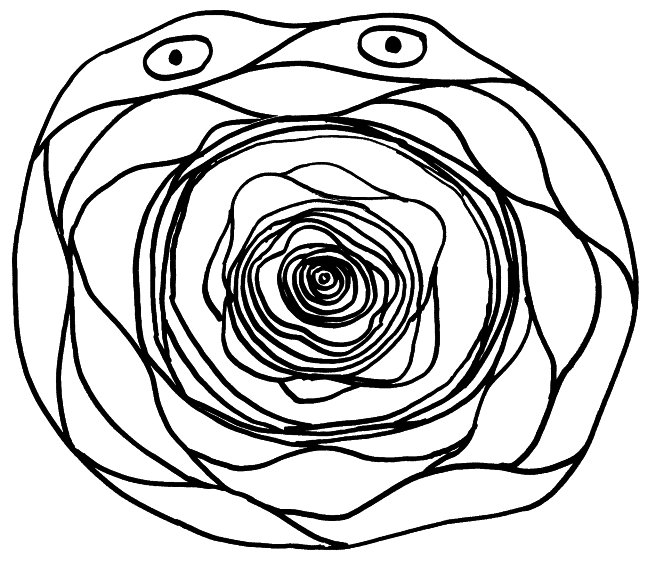
Des Rätsels Lösung: Das Oxytocin gelangt nicht in die Flüssigkeit zwischen den Zellen, weil es nicht ausgeschüttet wird, sondern nur bei direkten Zellkontakten zum Einsatz kommt. Die Oxytocin-produzierenden kortikalen Thymus-Epithelzellen, thymic nurse cells (TNC) oder Ammenzellen genannt, sind erheblich größer als die Thymozyten, binden diese mithilfe des Hormons eng an ihre Oberfläche und nehmen bis zu 50 (nach einigen Quellen sogar bis zu 200) Thymozyten im DP-Stadium vorübergehend in sich auf. Unter dem Mikroskop sieht man die kleinen Thymozyten oft geradezu bienenwabenartig in den Ammenzellen angeordnet (o.). Aus anderer Perspektive (u.) erkennt man, dass die Ammenzellen nur an einer Seite Thymozyten aufnehmen. Ob sie diese wirklich komplett in Vesikel, also Bläschen einschließen oder sie nur mit ihren Zellmembranlappen eng umarmen, ist umstritten.
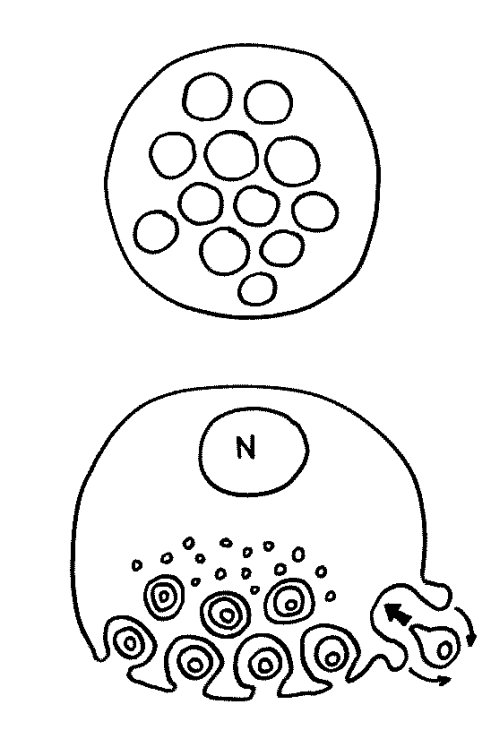
Klar ist dagegen, dass die Thymozyten nicht etwa passiv verschlungen werden, sondern aktiv an ihrer Aufnahme in die Ammenzellen teilnehmen: Wie in der Zeichnung unten rechts angedeutet, verformen sich die Thymozyten und bewegen sich auf die Ammenzelle zu, während die Membranausstülpungen der Ammenzelle immer länger werden und die Thymozyten umschließen.
Die Bezeichnung „Ammenzelle“ soll zum Ausdruck bringen, dass zumindest ein Teil der Thymozyten von diesem innigen Kontakt profitiert; sie werden von den Ammenzellen versorgt oder geschützt.

Dass Säugetierzellen andere Zellen desselben Organismus aufnehmen und später lebendig wieder ausscheiden können, statt sie zu verdauen, stieß in der Fachwelt auf ungläubiges Staunen. Außerdem war und ist die Funktion der Ammenzellen umstritten. Die mutmaßlich Oxytocin-gelenkte Kommunikation der beiden Zelltypen führte zunächst zu der Hypothese, dass die Ammenzellen unser Immunsystem mit unserem Hormonsystem verknüpfen und dafür sorgen, dass die T-Zellen später nicht Alarm schlagen, wenn sie im Körper mit Hormonen konfrontiert werden.
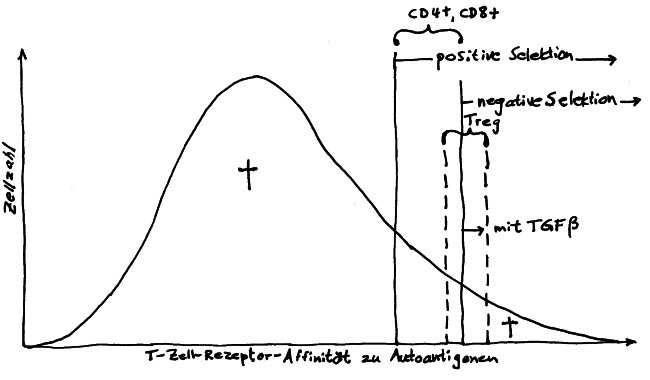
Diese Hypothese wurde zu einem Modell ausgebaut, demzufolge die Ammenzellen die internalisierten DP-Thymozyten generell auf die Stärke der Bindung zwischen T-Zell-Rezeptor und MHC-Antigen-Komplex prüfen und alle allzu bindungsfreudigen Thymozyten (in der nächsten Zeichnung links) absterben lassen. Schließlich sind alle Antigene, die den Thymozyten im Thymus auf einem MHC-Klasse-I- oder MHC-Klasse-II-Molekül präsentiert werden, notgedrungen Autoantigene, nämlich von körpereigenen Zellen selbst hergestellte Antigene.
Die Beseitigung der abgestorbenen Thymozyten scheinen Makrophagen zu übernehmen, die sich in der Nähe der Ammenzellen aufhalten und oft sogar im Inneren der Thymozyten-haltigen Höhlen in den Ammenzellen zu sehen sind, in denen sie womöglich ähnliche Dienste verrichten wie Putzerfische im Maul großer Raubfische. Nur Thymozyten, deren Rezeptoren zwar in der Lage sind, Autoantigen-beladene MHC-Moleküle zu erkennen, aber maßvoll darauf reagieren (rechts), werden intakt in die Freiheit entlassen.
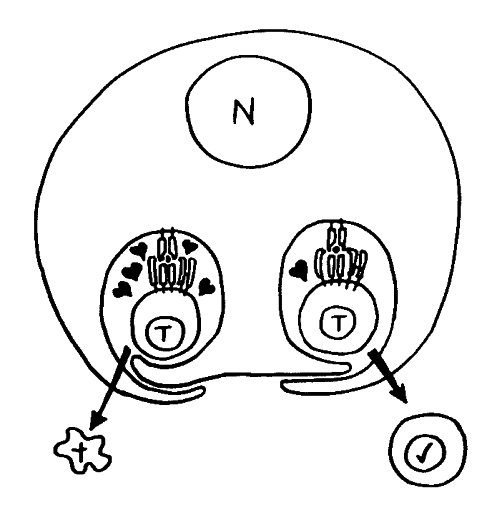
Demnach würden sich die Ammenzellen an der negativen Selektion beteiligen. Gegen dieses Modell spricht, dass die meisten Forscher Ammenzellen ausschließlich in der Thymusrinde finden, vor allem in deren Außenbereich direkt unter der Bindegewebskapsel, nicht aber an der Grenze zwischen Rinde und Mark oder gar im Mark, wo sich die negative Selektion nach vorherrschender Meinung abspielt. Manche Forscher widersprechen dem: Sie finden entweder auch im Mark Ammenzellen oder auch in der Rinde Indizien für eine negative Selektion. Aber auch sie sind der Ansicht, dass die Ammenzellen primär an der positiven Selektion beteiligt sind, die der negativen Selektion vorgeschaltet ist.
Ein japanisches Team (Y. Nakagawa et al.) brachte 2012 ein alternatives Modell ins Gespräch. Die Forscher hatten festgestellt, dass in den Ammenzellen DP-Thymozyten angereichert sind, die nach einer misslungenen ersten somatischen Rekombination ihrer Rezeptor-α-Kette eine weitere Neuanordnung dieser Gene durchlaufen haben. Die Ammenzellen wären demnach weder für die positive noch für die negative Selektion unentbehrlich, würden aber den Anteil der Thymozyten erhöhen, die die positive Selektion überleben.
Während der somatischen Rekombination der α-Ketten-Gene wird ein Modul V (für „variabel“) mit einem Modul J (für joint, also „Gelenk“) verbunden. Nur wenn das passgenau gelingt, kann von dem Gen eine Messenger-RNA abgelesen werden, die als Bauanleitung für die α-Kette dient. Häufig führt der erste Versuch zu einer aus dem Takt geratenen DNA-Sequenz, von der die Zelle keine brauchbaren Informationen mehr ablesen kann. Dann erhält die Zelle eine zweite, bei Bedarf auch eine dritte oder vierte Chance, bis schließlich alle V- und J- Module verbraucht sind oder aber ein intaktes Kombi-Modul entstanden ist.
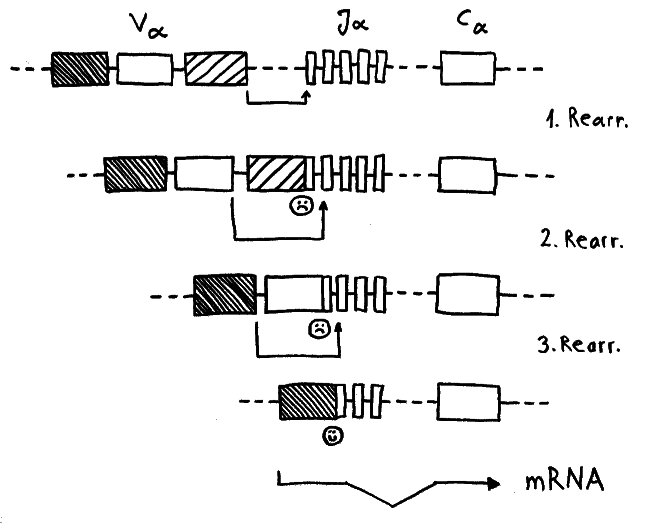
Dafür brauchen die Thymozyten offenbar einen Rückzugsort, an dem sie vor einem vorzeitigen Absterben geschützt sind. Die Höhlen in den Ammenzellen könnten dieser Ort sein: gewissermaßen Klosterzellen für Thymozyten, die sich in Ruhe neu erfinden wollen.

Warum solche Schutzräume nur bei der Neuanordnung der α-Ketten-Gene angeboten werden und nicht schon bei der somatischen Rekombination der β-Ketten, darüber kann ich nur wild spekulieren: Vielleicht hat der Organismus zu diesem Zeitpunkt schon so viel Energie und Zeit in die DP-Thymozyten investiert, dass es verschwenderisch wäre, sie nach dem ersten Fehlversuch absterben zu lassen. DN-Thymozyten könnten dagegen „billiger“, also bei einer gescheiterten β-Ketten-Rekombination mit weniger Aufwand zu ersetzen sein.
Mäuse, bei denen man durch genetische Eingriffe verhindert, dass ein Teil der kortikalen Thymus-Epithelzellen zu Ammenzellen heranreift, haben ein unauffälliges T-Zell-Profil; offenbar gelingt die Thymozyten-Reifung einschließlich der positiven und der negativen Selektion auch ohne Ammenzellen – vielleicht einen Hauch weniger effizient.
Diese vorläufige Lösung des Rätsels um die Ammenzellen ist zugegebenermaßen etwas unbefriedigend. Sie ist längst nicht allgemein anerkannt und kann schon morgen durch ein Revival des Selektionsmodells oder das Aufkommen eines dritten Modells überholt sein. Vor allem aber klingt sie arg unspektakulär angesichts der wildromantischen Verheißungen, die das Wort Oxytocin heraufbeschwört, und der Ungeheuerlichkeit, die das Verschlingen und Ausspeien lebender Zellen darstellt: Wenn Säugetierzellen ein so spektakulären Trick grundsätzlich beherrschen – wieso machen sich nicht häufiger von ihm Gebrauch?